-
If you are citizen of an European Union member nation, you may not use this service unless you are at least 16 years old.
-
You already know Dokkio is an AI-powered assistant to organize & manage your digital files & messages. Very soon, Dokkio will support Outlook as well as One Drive. Check it out today!
| |
Oral Pharmacological Agents for Type 2 Diabetes
Page history
last edited
by Robert Rushakoff, MD 13 years, 4 months ago
Oral Agents, Incretins and other "Non-Insulin" Pharmacologic Interventions for Diabetes
Last Author Revision: December 2010
INTRODUCTION
Diabetes has reached epidemic proportions in the United States (>23.6 million people or 7.8% of the population) as well as worldwide (>246 million people) and these numbers are projected to increase dramatically . Furthermore, the prevalence of insulin resistance, a major causative factor in the early development of type 2 diabetes and an independent risk factor for cardiovascular disease and the metabolic syndrome X, is even more widespread . This situation is further exacerbated by obesity, a major risk for developing type 2 diabetes. The number of adults overweight or obese in the US is 125 million (65% of population) and 1.3 billion worldwide . Since dietary modification and increased physical activity provide insufficient glucose control over the long-term course of the disease, the vast majority of patients require some type of pharmacological intervention .
In response to the enormity of the growing problem, efforts to identify and develop new pharmacological agents for type 2 diabetes have increased dramatically in recent years. These efforts have resulted in the successful introduction of several new treatment options, and additional new therapies will likely gain regulatory approval (US, Europe, Asia) in the near future. Currently, there are eight classes of oral pharmacological agents available to treat type 2 diabetes: sulfonylureas, meglitinides, metformin (a biguanide), thiazolidinediones, α-glucosidase inhibitors, Dipeptidyl Peptidase IV (DPP-IV) inhibitors, Bile Acid Sequestrants, and Dopamine Agonists. (Table 1). Besides the many forms of insulin (see chapter on insulin), there are also two classes of injectable medications currently used for treatment of diabetes: amylin analogue and GLP Agonists (glucagon like peptide agonist). Medications from these different classes of pharmaceutical agents may be used as treatment by themselves or more often a combination from multiple classes with multiple mechanisms of action is utilized. A variety of ‘fixed combination’ agents have been introduced (Table 2). These products may be useful to the patient as they provide two drugs in a single tablet, presumably offering convenience and increased compliance. They also enable patients to receive two medications for single medical insurance co-pay.
Table 1. Current Oral Pharmacological Therapies Used to Treat Type 2 Diabetes
|
Class
|
Brand
|
Manufacturer
|
Generic
Available
|
Daily Dose (mg)
|
|---|
|
Sulfonylureas
|
|
1st Generation
|
|
Chlorpropamide
|
none
|
generic
|
yes |
100-500 mg qd
|
| Tolazamide |
none |
generic |
yes |
100-750 mg qd or divided tid |
|
Tolbutamide
|
none |
generic |
yes |
500-2000 mg qd or divided bid
|
|
2nd Generation
|
|
Glyburide (Glibenclamide)
|
Diabeta
|
Aventis
|
yes |
2.5-10 mg qd or divided bid
|
|
Glyburide (Glibenclamide)
|
Micronase
|
Pharmacia
|
yes |
2.5-10 mg qd or divided bid
|
|
Glyburide (Glibenclamide)
|
Glynase
|
Pharmacia
|
yes |
0.75-12 mg qd or divided bid
|
|
Glipizide
|
Glucotrol
|
Pfizer
|
yes |
10 mg qd or divided bid
|
|
Glipizide
|
Glucotrol XL
|
Pfizer
|
yes |
5-10 mg qd
|
|
Glimepiride
|
Amaryl
|
Aventis
|
yes |
1-4 mg qd
|
|
Meglitinides
|
|
Repaglinide
|
Prandin
|
Novo Nordisk
|
no |
1.5-2 mg tid
|
|
Nateglinide
|
Starlix
|
Novartis
|
no |
60-120 mg tid
|
|
Biguanide
|
|
Metformin
|
Glucophage
|
Bristol-Myers Squibb
|
yes |
500-2500 mg qd-divided tid dosing
|
|
Metformin
|
Glucophage XR
|
Bristol-Myers Squibb
|
yes |
500-2000 mg qd
|
|
Thiazolidinediones
|
|
Rosiglitazone (restricted use in US)
|
Avandia
|
GlaxoSmithKline
|
no |
4-8 mg qd, 2-4 bid
|
|
Pioglitazone
|
Actos
|
Takeda/Eli Lilly
|
no |
15-45 mg qd
|
|
α-Glucosidase Inhibitors
|
|
Acarbose
|
Precose/Glucobay
|
Bayer, generics
|
yes |
50-100 mg tid
|
|
Miglitol
|
Glyset
|
Pharmacia
|
|
50-100 mg tid
|
| DDP-4 inhibitor |
|
Saxagliptin
|
Onglyza |
Bristol-Myers Squibb |
No |
2.5-5 mg qd |
| Sitagliptin |
Januvia |
Merck |
No |
25-100 mg qd |
| Bile Acid Sequestrant |
|
Colesevelam
|
WelChol
|
Daiichi Sankyo |
No |
1875 mg bid |
|
Dopamine Agonist
|
|
Bromocriptine
|
Parlodel
|
Novartis |
Yes |
0.25– 0.5 mg qd |
| Bromocriptine |
Cycloset |
Santarus |
(a quick release formulation) |
0.8-mg tabs - target range (1.6 - 4.8 mg) |
Table 2. Fixed Combination Therapies Treat Type 2 Diabetes
|
Drug 1
|
Drug 2
|
Brand
|
Manufacturer
|
Generic
Available
|
Available Doses
(mg Drug 1/mg Drug 2
|
|---|
|
Glyburide
|
Metformin
|
Glucovance
|
Bristol-Myers Squibb
|
yes |
1.25/250;2.5/500; 5/500
|
|
Glipizide
|
Metformin
|
Metaglip
|
Bristol-Myers Squibb
|
yes |
2.5/250; 2.5/500; 5/500
|
|
Glimepiride
|
Pioglitazone
|
Duetact
|
Takeda
|
no |
2/30; 4/30
|
|
Glimepiride
|
Rosiglitazone
|
Avandaryl
|
GlaxoSmithKline
|
no |
(restrected use in US)
1/4; 2/4; 4/4
|
| Sitagliptin |
Metformin |
Janumet |
Merck |
no |
50/500; 50/1000 |
| Saxagliptin |
Metformin |
Kombiglyze XR |
Bristol-Myers Squibb
|
no |
5 mg /500; 5/1000; 2.5/1000 |
|
Pioglitazone
|
Metformin
|
ACTOSPlusMet
|
Takeda
|
no |
15/500; 15/850
|
|
repaglinide
|
metformin |
PrandiMet |
Novo Nordisk |
no |
1/500; 2/500 |
|
Rosiglitazone
|
Metformin
|
Avandamet
|
GlaxoSmithKline
|
no |
(restricted use in US)
1/500; 2/500; 4/500; 2/1000; 4/1000
|
The actions of sulfonylureas and meglitinides involve the stimulation of insulin secretion; metformin suppresses hepatic glucose production; the thiazolidinedione class targets peripheral tissue insulin resistance; and the α-glucosidase inhibitors inhibit complex carbohydrate breakdown in the gut. A summary of their primary sites of action is given is Figure 2. There are no studies directly comparing the efficacy of all the oral agents. Data from multiple studies are provided in Tables 3 and 4, which summarize comparative efficacies when these drugs are used as monotherapy and in combination. A cost comparison of the medications is given in Table 5. Since several comprehensive reviews have focused on this topic , the overall objective of this chapter is to provide a concise, comparative overview of the available oral treatments, and to highlight some emerging approaches.
Table 3. Clinical Efficacy of Pharmacological Therapies to Treat Type 2 Diabetes When Used as Monotherapy or as Add On Treatment
| |
↓Fasting Plasma
Glucose
| | | |
|---|
|
Agent Class
|
(mg/dl)
|
(mmol/l)
|
↓HbA1C (%)
|
Insulin
|
Lipids
|
Body Weight
|
Major Side Effects
|
|---|
|
Note: caution must be taken in comparing the results presented above. Head to head comparisons of these agents are limited. The groups studied were often different , with significantly different baseline HgA1c levels. There is a strong correlation between baseline HbA1c and the magnitude of effectiveness of these agents (i.e. ability to decrease HbA1c). Significantly greater reductions in both fasting plasma glucose and HbA1c were observed in groups with higher baseline HbA1c. For those patients whose HbA1c was <8.0%, the reduction from therapy was only 0.1-0.2% (see below)
|
|---|
|
Sulfonylureas
|
60-70
|
3.3-3.9
|
0.8-2.0
|
Increase
|
No effect
|
Increase
|
Hypoglycemia
|
|
Meglitinides
|
65-75
|
3.6-4.2
|
0.5-2.0
|
Increase
|
No effect
|
Increase
|
Hypoglycemia
|
|
Biguanide (Metformin)
|
50-70
|
2.8-3.9
|
1.5-2.0
|
Decrease
|
↓TG↓LDL ↑HDL
|
Decrease
|
GI disturbances; LA1
|
|
Thiazolidinediones Pioglitazone
Rosiglitazone
|
60-80
|
3.3-4.3
|
1.4 -2.6
|
Decrease
|
↓ TG, -LDL
↑ HDL;
-TG, ↓
LDL,↑HDL
|
Increase
|
Fluid retention; decreased Hb, Congestive Heart Failure, fractures
|
|
α-Glucosidase inhibitors
|
25-30
|
1.9-2.2
|
0.7-1.0
|
No effect
|
No effect
|
No effect
|
GI disturbances
|
| DDP-4 inhibitor |
12-28 |
0.6.-1.5 |
0.5-0.8 |
increase |
No effect |
no effedt |
Risk of Pancreatitis (not proven) |
| Bile Acid Sequestrant |
15 |
.83 |
0.5 |
No effect |
-TG, ↓ LDL,
-HDL
|
No effect |
esophageal obstruction, bowel obstruction, fecal impaction, dysphagia,pancreatitis, nausea, constipation |
|
Dopamine Agonist
|
0-18 |
0-1.0 |
0.1-0.6 |
no effect |
no effect |
no effect |
Nasal Stuffiness, Nausea, headache, constrictive pericarditis, neuroleptic malignant syndrome, hypotension |
| GLP-1 agonist |
9-28 |
0.5-1.5 |
.7-.9 |
Increase |
no effect |
decrease |
Nausea, constipation, Risk of pancratitis (not proven) |
| Amylin |
|
|
|
|
|
decrease |
Nausea |
Table 4. Clinical Efficacy of Pharmacological Therapies to Treat Type 2 DiabetesWhen Used in Combination
|
Approved Combination
|
Baseline HbA 1C(%)
|
HbA 1C(%) a
|
Reference
|
|---|
|
aChange from baseline
bTroglitazone (this TZD no longer available) was added to patients failing on SU and metformin. Abbreviations: AGI, α-glucosidase inhibitor; MEG, meglitinide; Nat, nateglinide; Pio, pioglitazone; Rep, repaglinide; Rosi, rosiglitazone; SU, sulfonylurea; Tro, troglitazone; TZD, thiazolidinedione
|
|---|
|
SU + Metformin
|
8.8
|
-1.7
|
|
|
SU + Metformin
|
10.1
|
-1.0
|
|
|
SU + Metformin
|
11.0
|
-1.9
|
|
|
SU + Metformin
|
12.3
|
-3.3
|
|
|
SU + Metformin
|
10.8
|
-2.9
|
|
|
SU + TZD (Pio)
|
≥8.0
|
-1.3
|
|
|
SU + TZD (Rosi)
|
9.1
|
-0.9
|
|
|
SU + AGI (Acarbose)
|
7.4
|
-1.0
|
|
|
SU + AGI (Acarbose)
|
8.0
|
-0.9
|
|
|
SU + AGI (Acarbose)
|
9.0
|
-1.1
|
|
|
SU + AGI (Miglitol)
|
9.2
|
-0.5
|
|
|
Metformin + MEG (Rep)
|
8.3
|
-1.4
|
|
|
Metformin + MEG (Nat)
|
8.4
|
-1.5
|
|
|
Metformin + TZD (Pio)
|
≥8.0
|
-0.8
|
|
|
Metformin + TZD (Rosi)
|
8.9
|
-1.2
|
|
|
Metformin + AGI (Acarbose)
|
7.9
|
-0.8
|
|
|
Metformin + AGI (Acarbose)
|
8.5
|
-0.6
|
|
|
SU + Metformin + TZD (Tro) b
|
9.6
|
-1.4
|
|
Table 5. Cost Comparison of Pharmacological Therapies to Treat Type 2 Diabetes
|
Drug Class
|
Active Agent
|
Brand Name
|
Typical Daily Dose (mg)
|
Monthly Cost 1($ US)
|
|---|
|
1Estimated cost from Drugstore.com.
|
|---|
|
Sulfonylureas
|
Glyburide
|
Diabeta, Micronase
|
1.25-20
|
17-153
|
|
Sulfonylureas
|
Glyburide
|
generic
|
1.25-20
|
13-26
|
|
Sulfonylureas
|
Glyburide (micronized)
|
Glynase
|
1.5-12
|
24-109
|
|
Sulfonylureas
|
Glyburide (micronized)
|
generic |
1.5-12
|
5-12 |
|
Sulfonylureas
|
Glipizide
|
Glucotrol
|
5-20
|
20-67
|
|
Sulfonylureas
|
Glipizide
|
generic |
5-20
|
6-12 |
|
Sulfonylureas
|
Glipizide XL
|
Glucotrol XL
|
5-20
|
25-80
|
|
Sulfonylureas
|
Glipizide XL
|
generic |
5-20
|
19-40 |
|
Sulfonylureas
|
Glimepiride
|
Amaryl
|
1-4
|
30-62
|
|
Sulfonylureas
|
Glimepiride
|
generic |
1-4
|
13-15 |
|
Meglitinides
|
Repaglinide
|
Prandin
|
1.5-6
|
192
|
|
Meglitinides
|
Nateglinide
|
Starlix
|
180-360
|
147-199
|
|
Biguanide
|
Metformin
|
Glucophage
|
500-2000
|
35-141
|
|
Biguanide
|
Metformin
|
generic |
500-2000
|
6-32 |
|
Biguanide
|
Metformin xr
|
Glucophage XR
|
500-2000
|
35-140
|
|
Biguanide
|
Metformin xr
|
generic |
500-2000
|
6-24 |
|
Thiazolidinediones
|
Rosiglitazone
|
Avandia
|
4-8
|
130-240
|
|
Thiazolidinediones
|
Pioglitazone
|
Actos
|
15-45
|
150-234
|
|
α-Glucosidase Inhibitors
|
Acarbose
|
Precose/ Glucobay
|
150-300
|
88-102
|
|
α-Glucosidase Inhibitors
|
Acarbose
|
generic |
150-300
|
90-90
|
|
α-Glucosidase Inhibitors
|
Miglitol
|
Glyset
|
150-300
|
88-100
|
| DDP-4 inhibitor |
Sitagliptin |
Januvia |
100 |
193 |
| DDP-4 inhibitor |
Saxagliptin |
Onglyza |
2.5-5 |
189 |
| Bile Acid Sequestrant |
colesevelam |
Welchol |
3750 |
224 |
|
Dopamine Agonist
|
Bromocriptine |
|
0.25– 0.5 |
62-130 |
| GLP-1 agonist |
Exenatide |
Byetta |
5-10 mcg SQ bid |
245-271 |
| GLP-1 agonist |
Liraglutide |
Victoza |
1.2-1.8 mg SQ qd |
|
| Amylin |
Pramlintide |
Symlin |
15-120 mcg |
300 |
| |
|
|
|
|
Treatment Goals
It is well established that, in poorly-controlled individuals with diabetes, both macrovascular and especially microvascular complications are increased . Elevated post-prandial glucose is also associated with an increased risk for the development of macrovascular disease . In the United Kingdom Prospective Diabetes Study (UKPDS) a reduction of HbA1c significantly decreased the risk for the development of microvascular complications in patients with type 2 diabetes . Accordingly, recommendations for treatment goals have been proposed by several professional organizations. The American Diabetes Association recommends an HbA1c of less than 7% . Since there was some evidence that increased risk for the develpment of cardiovascular disease begins at concentrations of HbA1c in the normal range, the American Association of Clinical Endocrinologists recommends an HbA1c of 6.5% or less . There more ambitious goals may be tempered with the release of three large randomized prospective studies showing lowering glucose levels to HgA1c below 7% did not reduce the risk of cardiovascular disease.
Though a majority of diabetic patients are being treated, many are unable to achieve the currently recommended goal of HbA1c <6.5% (AACE) or <7% (ADA). Bloomgarden et al reported results from a meta-regresion analysis of 61 clinical trials evaluating the efficacy of the five major classes of oral antihyperglycemic agents. His results indicate that there is a strong correlation between baseline HbA1c and the magnitude of effectiveness of these agents (i.e. ability to decrease HbA1c). Significantly greater reductions in both fasting plasma glucose and HbA1c were observed in groups with higher baseline HbA1c. For those patients whose HbA1c was <8.0%, the reduction from therapy was only 0.1-0.2% . These results are presented graphically in Figure 3. Thus, expectations for the overall magnitude of effect from a given agent might be overly optimistic when treating patients whose baseline HbA1c is <7.5-8.0%. These observations certainly reinforce the need to use combination therapy (dual, triple, or even four agents) to achieve a recommended target for HbA1c. In addition, these results point out that comparing efficacy between drugs may be difficult when the HgA1c baseline is different in studies.
Sulfonylureas
Sulfonylureas, derived from sulfonic acid and urea, were initially developed in the 1950’s and have remained a cornerstone of therapy for type 2 diabetes . The combination of their proven efficacy in most patients, low incidence of adverse events, and low cost has contributed to their success and continued use. They are frequently classified as either 1st generation or 2nd generation agents. First generation sulfonylureas (acetohexomide, chlorpropamide, tolazamide, and tolbutamide) possess a lower binding affinity for the ATP-sensitive potassium channel, their molecular target (vide infra), and thus require higher doses to achieve efficacy, increasing the potential for adverse events. In addition, the plasma half-life of 1st generation sulfonylureas is extended (e.g. 5-36 h) compared to the 2nd generation agents. Chlorpropamide was once the most commonly used oral agent, but now it is rarely prescribed. Unique complications associated with chlorpropamide are hyponatremia (SIADH) and an alcohol flushing reaction (disulfiram-Antibuse reaction). In addition, tolbutamide, acetohexamide and tolazamide generally require 2 or 3 doses per day and are rarely used.
More recently, 2nd generation sulfonylureas including glyburide (glibenclamide; , glipizide and glimepiride were introduced, and are now widely used. The 2nd generation sulfonylureas are much more potent compounds (~ 100-fold), possess a more rapid onset of action, and generally have shorter plasma half-lives and longer duration of action compared to the 1st generation agents.
Figure Structures of Glyburide, Glipizide and Glimepiride

Mechanism of Action
Sulfonylureas are insulin secretogogues, since they control blood glucose levels by directly stimulating first-phase insulin secretion in the pancreatic β cells. Through the concerted efforts of GLUT2 (the high Km glucose transporter) , glucokinase (the glucose sensor) , and glucose metabolism, these cells are responsible for sensing and secreting the appropriate amount of insulin in response to a glucose stimulus . Mitochondrial glucose metabolism leads to ATP generation and increases the intracellular ratio of ATP/ADP, which results in the closure of the ATP-sensitive potassium channel (KATP; a 140 kDa membrane protein) on the plasma membrane. Closure of this channel depolarizes the membrane and triggers the opening of voltage-sensitive calcium channels, leading to the rapid influx of calcium. Increased intracellular calcium causes an alteration in the cytoskeleton, and stimulates translocation of insulin-containing secretory granules to the plasma membrane and the exocytotic release of insulin.
The KATP channel is comprised of two subunits, both of which are required for the channel to be functional. One subunit contains the cytoplasmic binding sites for both sulfonylureas and ATP, and is designated as the sulfonylurea receptor type 1 (SUR1). The other subunit is the potassium channel, which acts as the pore-forming subunit
. Either an increase in the ATP/ADP ratio or ligand binding (by sulfonylureas, meglitinides) to SUR1 results in the closure of the KATP channel and insulin secretion. Studies comparing sulfonylureas and non-sulfonylurea insulin secretogogues have identified several distinct binding sites on the SUR1 that cause channel closure. Some sites exhibit high affinity for glyburide and other sulfonylureas, while other sites exhibit high affinity for the non-sulfonylurea secretogogues (vide infra).
Efficacy
The over clinical efficacy of sulfonylureas in patients with type 2 diabetes is related to the pre-treatment levels of fasting plasma glucose and HbA1C. The higher the fasting glucose level, the greater the effect will be. In patients with a pre-treatment glucose level of approximately 200 mg/dl (11.1 mmol/l), sulfonylureas typically will reduce glucose by 60-70 mg/dl (3.3-3.9 mmol/l) and HbA1C by 1.5-2% (Table 3). The most responsive patients are those who exhibit mild-to-moderate fasting hyperglycemia (<200-240 mg/dl; <12.2-13.3 mmol/l), along with adequate residual β-cell function (evidenced by elevated fasting C-peptide). When used at maximally effective doses, results from well-controlled clinical trials have not indicated a superiority of one 2nd generation sulfonylurea over another. Similarly, 2nd generation sulfonylureas exhibit similar clinical efficacy compared to the 1st generation agents. The principal advantage of glimepiride and Glucotrol XL compared to other agents is the once daily dosing regimen. Approximately 10-20% of patients will exhibit a poor initial response to sulfonylureas (primary failures). While these patients are typically those who have severe fasting hyperglycemia (>280 mg/dl; >15.5 mmol/l) and reduced fasting C-peptide levels, these tests are not specific enough to help decide on the usefulness of a sulfonylurea for an individual patient. In addition, treatment with sulfonylureas results in the eventual loss of therapeutic effectiveness (secondary failure) in the range of 3-10% per year .
The major side effect from sulfonylurea treatment is hypoglycemia. This side effect is really just an extension of the therapeutic objective. Mild hypoglycemic events occur in approximately 2-4% of patients and severe hypoglycemic reactions that require hospitalization occur at a frequency of 0.2-0.4 cases per 1000 patient-years of treatment . In light of this, initiation of treatment with sulfonylureas should be at the lowest recommended dose. An additional undesirable effect of sulfonylurea therapy (as is also the case with insulin therapy) is weight gain. In the UKPDS, sulfonylurea treatment caused a net weight gain of 3 kg, which occurred during the first 3-4 years of treatment and then stabilized . In contrast, weight gain in response to insulin therapy increased progressively for the duration of the study. As mentioned above, chlorpropamide is associated with hyponatremia (SIADH) and an alcohol flushing reaction (disulfiram-Antibuse reaction). All the agents can cause intrahepatic cholestasis. Rarely maculopapular or urticarial rashes occur.
In renal failure, the dose of the sulfonylurea agent will require adjustment based on glucose monitoring. The half-life of insulin is extended in renal failure and thus there is an increased risk for hypoglycemia. This risk is typically manifest with fasting hypoglycemia.
Meglitinides: Repaglinide and Nateglinide
The meglitinides are a novel class of non-sulfonylurea insulin secretogogues characterized by a very rapid onset and abbreviated duration of action. Repaglinide (Prandin®), a benzoic acid derivative introduced in 1998, was the first member of the meglitinide class. Nateglinide (Starlix®) is a derivative of the amino acid D-phenylalanine and was introduced to the market in 2001. Unlike sulfonylureas, repaglinide and nateglinide stimulate first-phase insulin release in a glucose-sensitive manner, theoretically reducing the risk of hypoglycemic events. The delivery of insulin as an early, transient ‘burst’ at the initiation of a meal affords several major physiological benefits (reviewed in ). These include rapidly suppressing hepatic glucose production and reducing the stimulus for additional insulin that would be required subsequently to dispose of a larger glucose load. Thus, the rapid onset/short duration stimulation of insulin release by meglitinides should enhance control of prandial hyperglycemia, while reducing the risk for post-absorptive hypoglycemia and limiting exposure to hyperinsulinemia.
Figure Structure of Nateglinide and Repaglinide

Mechanism of Action
Similar to sulfonylureas, meglitinides are insulin secretogogues, since they control blood glucose levels by directly stimulating first-phase insulin secretion in the pancreatic β cells. Receptor-binding studies performed in βTC-3 cells identified a high-affinity repaglinide (KD = 3.6 nmol/l) site having lower affinity for glyburide (14.4 nmol/l), and one high-affinity glyburide (25 nmol/l) site having lower affinity for repaglinide (550 nmol/l)(Figure 1.4) . Repaglinide is approximately 5 times more potent than glyburide in stimulating insulin secretion. Unlike glyburide (and other sulfonylureas), repaglinide does not stimulate insulin secretion in vitro in the absence of glucose. Rather, it enhances glucose-stimulated insulin secretion especially at 180 mg/dl (10 mmol/l) glucose.
The mechanism of action of nateglinide (Figure 1.5) also involves the binding to and closure of the KATP channel resulting in membrane depolarization, an influx of calcium, and insulin exocytosis . The kinetics of interaction of nateglinide with the KATP channel are distinct compared to both rapaglinide and sulfonylureas, and accounts for its rapid insulinotropic effects. The onset of action of nateglinide is similar to that of glyburide but three-fold more rapid than that of rapaglinide
. When nateglinide is removed from the KATP channel, its effect is reversed twice as quickly as glyburide and five times more quickly than rapaglinide. Thus, nateglinide initiates a more rapid release of insulin that is shorter in duration compared to rapaglinide , despite having an in vivo pharmacokinetic profile that is similar .
Efficacy
The efficacy of rapaglinide, when used as a monotherapy, is similar to sulfonylureas . Repaglinide treatment of patients with type 2 diabetes reduced fasting plasma glucose by approximately 60 mg/dl and HbA1C by 1.7% . In a double-blind placebo-controlled study, rapaglinide had similar effects on lowering HbA1C (0.5-2%) and fasting plasma glucose (65-75 mg/dl; 3.6-4.2 mmol/l) compared to glyburide . Repaglinide is also efficacious when used in combination with either metformin or troglitazone (a thiazolidinedione withdrawn from the market). In patients treated with rapaglinide and metformin, HbA1C was decreased from 8.3% to 6.9% and fasting plasma glucose by 40 mg/dl (2.2 mmol/l) . Although lowered, the changes observed in subjects treated with either repaglinide or metformin monotherapy were not significant for HbA1C (0.4 and 0.3% decrease, respectively), or fasting plasma glucose (9 mg/dl (0.5 mmol/l) increase and 5.4 mg/dl (0.3 mmol/l) decrease, respectively). Significant increases in body weight occurred in the both repaglinide and combined therapy groups (2.4 ± 0.5 and 3.0 ± 0.5 kg, respectively).
The combination therapy of rapaglinide and troglitazone showed a significant reduction in mean HbA1C values (1.7%) that was greater than with either type of monotherapy
. Repaglinide monotherapy resulted in a reduction of HbA1C values that was significantly greater than troglitazone (0.8% vs. 0.4%). In addition, combination therapy was more effective in reducing fasting plasma glucose (80 mg/dl) than either repaglinide (43 mg/dl) or troglitazone (46 mg/dl) monotherapies. Repaglinide is also efficacious when used in combination with other available thiazolidinediones, rosiglitazone (Avandia®) and pioglitazone (Actos).
The efficacy of nateglinide when used as a monotherapy is similar to sulfonylureas and repaglinide (29;40). However, several therapeutically attractive features distinguish nateglinide from repaglinide and sulfonylureas. Nateglinide produces a more rapid post-prandial increase in insulin secretion, and its duration of response is shorter than that of glyburide . Thus, the risk of post-absorptive hypoglycemia should be lower than with either sulfonylureas or rapaglinide, but this has not been demonstrated in studies to date.
The efficacy of nateglinide treatment has been evaluated alone and in combination with metformin in patients with type 2 diabetes . In this randomized double-blind study, patients with an HbA1C level between 6.8 and 11.0% during a 4-week placebo run-in received 24 weeks’ treatment with 120 mg nateglinide before meals (n = 179), 500 mg metformin three times a day (n = 178), combination therapy (n = 172), or placebo (n = 172). At the study conclusion, HbA1C and fasting plasma glucose were significantly reduced from baseline with nateglinide [0.5% and 12.6 mg/dl (0.7 mmol/l), respectively] and metformin [0.8% and 28.8 mg/dl (1.6 mmol/l), respectively], but was increased with placebo [0.5% and 7.2 mg/dl (0.4 mmol/l), respectively]. Combination therapy was additive [HbA1C, 1.4% and glucose, 43.2 mg/dl (2.4 mmol/l)]. Although only preliminary data are available, nateglinide also appears effective when used in combination with thiazolidinediones .
In a direct comparison of Repaglinide and Nateglinide, the known pharmacodynamic differences in the drugs are evident on the clinical outcomes. The longer acting Repaglinide has significant effects on fasting glucose levels while Nateglinide does not. (figure 3a) These clinical findings are important and can be incorporated into clinical decision making. For example, if the main issue for the patient is postprandial hyperglycemia, and fasting glucoses are near normal, an agent that has limited effect on the fasting glucose would be beneficial.

Figure 3a. Comparison of Repaglinide and Nateglinide
Side Effects
In 1-year trials, the most common adverse events reported in repaglinide recipients (n = 1,228) were hypoglycemia (16%), upper respiratory tract infection (10%), rhinitis (7%), bronchitis (6%) and headache (9%). The overall incidence of hypoglycemia was similar to that recorded in patients receiving glibenclamide, glipizide or gliclazide (18%; n = 597); however, the incidence of serious hypoglycemia appears to be slightly higher in sulphonylurea recipients. Weight gain does occur in patients treated with repaglinide, but the magnitude is significantly less compared to treatment with glyburide. In patients switched from sulfonylureas to repaglinide, no weight gain was observed; in drug-naïve patients, repaglinide-treatment increased body weight by approximately 3% (6lb) .
The clinical trials of nateglinide carried out to date have found the drug to be safe and well tolerated. Dosage regimens ranging from 60 to 240 mg have been evaluated. The most common adverse effects are nausea, diarrhea, dizziness, and lightheadedness. The incidence of mild hypoglycemia is lower with nateglinide than for rapaglinide and no reports of severe hypoglycemia, consistent with the mechanism of action of nateglinide. In the clinical studies carried out to date, there have been no reports of any increase body weight gain.
Metformin
Metformin (dimethlybiguanide; Glucophage®) is a synthetic analog of the natural product guanidine, whose history as a treatment for diabetes can be traced to medieval times . Metformin has surpassed the sulfonylureas as the most prescribed oral agent for type 2 diabetes in the US. In the major European markets, metformin is the second most prescribed agent after glyburide . The widespread acceptance of metformin evolved after the realization that lactic acidosis was not a major problem in individuals with normal renal function. Phenformin, a structurally similar analog of metformin, was previously withdrawn from the market in many countries due its propensity to induce lactic acidosis. Metformin is recommended as a first-line therapy in newly diagnosed individuals, and can be used in combination with an insulin secretagogue (sulfonylurea or meglitinide), thiazolidinedione, α-glucosidase inhibitor, exenatide, DPP-4 inhibitor or insulin . When used as a monotherapy, metformin decreases HbA1c by 1.5-2.0%, increases insulin sensitivity, does not promote weight gain, and has an acceptable side effect profile.
Figure Structure of Metformin

Mechanism of Action
An elevated rate of basal hepatic glucose output is the primary determinant of elevated fasting blood glucose levels in patients with type 2 diabetes . The primary effect of metformin (Figure 1.6) is the suppression of basal hepatic glucose production, thereby reducing fasting plasma glucose . Despite the large number of studies both in vitro and in humans that have established this mode of action, the molecular target of metformin action still awaits identification. Metformin does not stimulate insulin secretion; in contrast, metformin reduces fasting plasma insulin and improves whole-body insulin-stimulated glucose metabolism (insulin sensitivity) . While it is possible that the beneficial effect of metformin on insulin sensitivity is mediated directly, a more likely explanation is that it is secondary to a reduction in hyperglycemia, triglycerides, and free fatty acids.
Recent in vitro and in vivo evidence has shown that metformin activates the AMP-activated protein kinase (AMPK) , a major cellular regulator of lipid and glucose metabolism . As a result, acetyl-CoA carboxylase activity was reduced, fatty acid oxidation induced (due to decreased malonyl-CoA), and the expression of lipogenic enzymes along with SREBP-1, a key lipogenic transcription factor, suppressed . The use of a novel AMPK inhibitor indicated that AMPK activation was required for the inhibitory effect of metformin on glucose production in hepatocytes. In isolated rat skeletal muscles, metformin stimulated glucose uptake coincident with AMPK activation. These results are intriguing in that they implicate the activation of AMPK as a unified explanation for the beneficial effects of metformin.
Efficacy
A large number of well-controlled clinical studies have established that metformin monotherapy consistently reduces fasting plasma glucose by 60-70 mg/dl (3.3-3.9 mmol/l) and HbA1c by 1.5-2.0% . Thus, the efficacy of metformin is in the same range as that observed for monotherapy treatment with sulfonylureas. Similar to the sulfonylurea treatment, the overall magnitude of response to metformin is directly related to the starting fasting plasma glucose concentration. Metformin also reduces fasting plasma insulin, triglycerides, and free fatty acids . Unlike sulfonylurea treatment, metformin monotherapy is not associated with weight gain and even promotes a modest weight loss. When used in combination with other oral agents or insulin, weight gain is not observed. Metformin is the only oral agent that when used as a monotherapy has been reported to reduce the risk of developing macrovascular complications .
Historically, when patients with type 2 diabetes in the US had mean HbA1c of approximately 10% and fasting plasma glucose approximately 200-240 mg/dl, in patients of this sort, monotherapy with either metformin or a sulfonylurea generally decreased plasma glucose to <140 mg/dl (<7.8 mmol/l) in about 25-30% of patients. In contrast, combined metformin and sulfonylurea therapy increased the percentage of patients who achieve this level of control to approximately 60-70% . When added to a sulfonylurea, the effects of both agents are additive, consistent with their different mechanisms of action. Interestingly, in patients that no longer responded to sulfonylurea treatment (secondary failures) and were removed from treatment, addition of metformin had minimal effects . Thus, in these patients, sulfonylurea treatment was still eliciting an effect, emphasizing the need to continue treatment with both agents.
The additive effect of metformin and sulfonylurea therapy is illustrated in Figure 4. As shown, there was no change in glucose levels when the sulfonylurea was changed to metformin. However, when the metformin was added, there was a dramatic decrease in plasma glucose. In fact, this pattern is seen in virtually all studies comparing two oral agents. This concept is illustrated in Figure 5. When a patient is on drug A and they are changed to drug B, no improvement in glucose control will be seen. However, if drug B is added to drug A, there is an improvement. This concept can often be extended by the addition of drug C, drug D, etc.
Benefits Beyond Glucose Control
Cardiovascular Disease
In a retrospective study on the effect of metformin-containing antidiabetic regimens on all-cause mortality in veterans with Type 2 Diabetes Mellitus, there was a decreased hazard ratio for all cause mortality for patients taking metformin whether they were also on other oral agents (0.77 p<0.01 )or on insulin (0.62 p<0.04). () In the United Kingdom Prospective Diabetes Study, 10 years after the study was completed, if the subjects had been on treatment regimens that included metformin there was a reduction in any diabetes-related end point (21%, P=0.01) myocardial infarction (33%, P=0.005) and death from any cause (27%, P=0.002). () In the REACH Registry (Reduction of Atherothrombosis for Continued Health) 19,691 patients with diabetes and established atherothrombosis were assessed for the effect of metformin use in circumstances when the medication would historically be contraindicated. They found the mortality rates were 6.3% (95% confidence interval [CI], 5.2%-7.4%) with metformin and 9.8% 8.4%-11.2%) without metformin; the adjusted hazard ratio (HR) was 0.76 (0.65-0.89; P<.001). Association with lower mortality was consistent among subgroups, noticeably in patients with a history of congestive heart failure (HR, 0.69; 95% CI, 0.54-0.90; P=.006), patients older than 65 years (0.77; 0.62-0.95; P=.02), and patients with an estimated creatinine clearance of 30 to 60 mL/min/1.73 m2 (0.64; 95% CI, 0.48-0.86; P=.003) (to convert creatinine clearance to mL/s/m2, multiply by 0.0167). ()
Side Effects and Contraindications
The most common side effects of metformin are gastrointestinal disturbances (abdominal discomfort, diarrhea), which occur in approximately 20-30% of patients. These effects are generally transient, and can be minimized or avoided by careful dose titration. The incidence of lactic acidosis is rare and occurs with a frequency of 3 cases per 100,000 patient-years. It appears that •Metformin probably not as unsafe as previously thought. 25% of users have relative contraindication and yet lactic acidosis remains rare. In addition, the patients who do develop lactic acidosis usually have acute renal failure and previously had normal renal function.
However, metformin is contraindicated in patients with impaired renal function and liver disease.
The risk of lactic acidosis can be minimized when the following are considered:
-
Withhold in conditions predisposing to renal insufficiency and/or hypoxia
-
CV collapse
-
Acute MI or acute CHF
-
Severe infection
-
Use of iodinated contrast material
-
Major surgical procedures
-
Officially, Metformin should not be prescribed for patients with:
-
Renal dysfunction [e.g. Scr >1.5 mg/dl (males), >1.4 mg/dl (females) or abnormal CrCl)
-
Liver dysfunction
-
History of alcohol abuse/binge drinking
-
Acute or chronic metabolic acidosis
With the introduction of the eGFR (estimated glomerular filtration rate) the question of a cutoff value for using this test for discontinuing metormin has been raised. An eGFR of 36 ml/min per 1.73m2 would be somewhat neutral to current use. Use of a novel assay for metformin levels has confirmed that metformin levels are not increased with eGFR levels as low as 30 ml/min per 1.73m2 .
Vitamin B12 deficiency: Both short and long term use of metformin has been found to decrease vitamin B12 levels. (decreased 4.2-47%). Metformin is thought to induce malabsorption of vitamin B12 and intrinsic factor in the ileum, an effect that can be reversed by increased calcium intake. Anemia may be minimal to severe (RR author observations). However, metformin associated vitamin B12 deficiency may present without anemia and only as a peripheral neuropathy, possibly being misdiagnosed as diabetic neuropathy. It has been speculated that exhaustion of vitamin B12 stores usually occurs after twelve to fifteen years of absolute and as metformin has been available in the United States for approximately fifteen years these clinical findings may now be coming to light.
Thiazolidinediones: Pioglitazone and Rosiglitazone
Pioglitazone (Actos®) and rosiglitazone (Avandia®) are members of the thiazolidinedione class of insulin sensitizing compounds originally discovered and characterized for their glucose- and lipid-lowering activity . These compounds decrease insulin resistance and enhance the biological response to endogenously produced insulin, as well as insulin administered by injection . Until September 23, 2010, each drug was approved for use in the US as monotherapy, which results in a significant reduction in fasting plasma glucose by 60-80 mg/dl and in HbA1c by 1.4-2.6% . In addition, pioglitazone is approved for use in combination with insulin, metformin, or a sulfonylurea, and rosiglitazone is approved for use in combination with metformin or a sulfonylurea. Troglitazone (Rezulin®), another member of this chemical class, was withdrawn from US, European, and Japanese markets in 2000 due to idiosyncratic hepatic reaction leading to hepatic failure and death in some patients. Although there are some data from animal studies suggesting that hepatic toxicity might be characteristic of the thiazolidinedione class , current clinical evidence indicates that pioglitazone and rosiglitazone treatment do not result in liver toxicity . As of September 2010, as a result of cardiovascular concerns (see below) Rosiglitazone was removed from the European market and use in the United States was restricted by the FDA. Use of rosiglitazone was restricted to patients who could not use any other medication. In addition, the patient must be informed about the potential cardiovascular risks.
Figure Structures of Rosiglitazone and Pioglitazone

Mechanism of Action
The primary effects of pioglitazone (Figure 1.7) and rosiglitazone (Figure 1.8) are the reduction of insulin resistance and improvement of insulin sensitivity, resulting in a reduction of fasting plasma glucose, insulin, and free fatty acids . Unlike other existing anti-diabetic medications that possess a very rapid onset of activity, pioglitazone and rosiglitazone exhibit a characteristic delay from 4-12 weeks in the onset of their therapeutic benefits. This is likely related to their mode of action, which involves the regulation of gene expression . Pioglitazone and rosiglitazone are selective agonists for the peroxisome proliferator-activated receptor γ (PPARγ), a member of the superfamily of nuclear hormone receptors that function as ligand-activated transcription factors . The PPAR family, which also includes PPARα and PPARδ, functions as receptors for fatty acids and their metabolites (e.g. eiconasoids) and, consequently, plays a critical physiological role the regulation of glucose, fatty acid, and cholesterol metabolism. PPARα is the receptor for the fibrate class of lipid-lowering drugs, and PPARδ is involved in the regulation of high-density lipoprotein metabolism .
The structure-activity relationship between PPARγ agonists and their glucose lowering activity in vivo has been established (72). In the absence of ligand, PPARs bind as heterodimers with the 9-cis retinoic acid receptor (RXR) and a multi-component co-repressor complex to a specific response element (PPRE) within the promoter region of their target genes . Once PPAR is activated by ligand, the co-repressor complex dissociates allowing the PPAR-RXR heterodimer to associate with a multi-component co-activator complex resulting in an increased rate of gene transcription. The target genes of PPARγ include those involved in the regulation of lipid and carbohydrate metabolism .
It does not appear that rosiglitazone and pioglitazone improve insulin sensitivity and glucose disposal by direct effects on either liver or muscle. PPARγ is expressed chiefly in adipose tissue, and its expression in liver and skeletal muscle is low . Thus, it is more likely that the primary effects of these drugs are on adipose tissue, followed by secondary benefits on other target tissues of insulin . The ability of rosiglitazone and pioglitazone to decrease circulating free fatty acids could lead to an improvement in insulin action in the periphery . More recently, PPARγ agonists have been reported to increase the expression and circulating level of adiponectin (Acrp30), an adipocyte-derived protein with insulin sensitizing activity , in diabetic rodents and in patients with type 2 diabetes . Recognition of the importance of PPARγ in the overall regulation of carbohydrate and lipid metabolism along with growing realization that the adipocyte is an endocrine organ suggests that investigations in this area will intensify, and perhaps uncover additional mechanisms by which rosiglitazone and pioglitazone improve insulin sensitivity and glucose disposal.
Efficacy
Rosiglitazone
The clinical efficacy of rosiglitazone and pioglitazone therapy has been extensively reviewed . Two 26-week, double blind, placebo-controlled clinical studies have established that rosiglitazone monotherapy reduces fasting plasma glucose and HbA1c in patients with type 2 diabetes . Treatment with rosiglitazone at 4 mg/day reduced fasting plasma glucose by approximately 30-45 mg/dl and HbA1c by 0.8-1.0%, compared with placebo. Treatment with rosiglitazone at 8 mg/day reduced fasting plasma glucose by approximately 45-65 mg/dl and HbA1c by 1.1-1.5%, compared with placebo (10;57;87). In patients with type 2 diabetes inadequately controlled with metformin, rosiglitazone produced a significant reduction in HbA1c compared to metformin treatment alone (94;95). In another study in which rosiglitazone was directly compared to a maximum stable dose of glyburide (15 mg/day), rosiglitazone reduced fasting plasma glucose by 25 mg/dl at 4 mg/day, and 40 mg/dl at 8 mg/day . The reduction in HbA1c was 0.7% for glyburide, 0.3% for rosiglitazone at 4 mg/day, and 0.5% for rosiglitazone at 8 mg/day.
Pioglitazone
Double blind, placebo-controlled studies with pioglitazone as monotherapy, have established that this agent reduces fasting plasma glucose HbA1c in patients with type 2 diabetes . Patients treated with 15, 30, or 45 mg (once daily) pioglitazone had significant mean decreases in HbA1c (range -1.00 to -1.60% difference from placebo) and fasting plasma glucose (-39.1 to -65.3 mg/dl difference from placebo). The decreases in fasting plasma glucose were observed as early as the second week of therapy; maximal decreases occurred after 10-14 weeks and were maintained until the end of therapy (week 26). There was no evidence of drug-induced hepatotoxicity, or elevated alanine aminotransferase activity.
The efficacy and tolerability of pioglitazone in combination with metformin has been assessed in patients with type 2 diabetes mellitus . Patients receiving pioglitazone (30 mg) + metformin had statistically significant mean decreases in HbA1c (-0.83%) and fasting plasma glucose levels (-37.7 mg/dl) compared with placebo + metformin. Decreases in fasting plasma glucose levels occurred as early as the fourth week of therapy, the first time point at which fasting plasma glucose was measured. The pioglitazone + metformin group had significant mean percentage changes in levels of triglycerides (-18.2%) and high-density lipoprotein cholesterol (+8.7%) compared with placebo + metformin. Mean percentage increases were noted in low-density lipoprotein cholesterol levels (7.7%, pioglitazone + metformin; 11.9%, placebo + metformin) and total cholesterol (4.1%, pioglitazone + metformin; 1.1%, placebo + metformin), with no significant differences between groups. In the extension study, patients treated with open-label pioglitazone + metformin for 72 weeks had mean changes from baseline of -1.36% in HbA1c and -63.0 mg/dl in fasting plasma glucose. In this study, there was no evidence of drug-induced hepatotoxicity.
The efficacy and tolerability of pioglitazone in combination with a sulfonylurea has been also assessed in patients with type 2 diabetes mellitus . Patients receiving pioglitazone (30 mg) + metformin had statistically significant mean decreases in HbA1c (-0.83%) and fasting plasma glucose levels (-37.7 mg/dl) compared with placebo + metformin. Decreases in fasting plasma glucose levels occurred as early as the fourth week of therapy, the first time point at which fasting plasma glucose was measured. The pioglitazone + metformin group had significant mean percentage changes in levels of triglycerides (-18.2%) and high-density lipoprotein cholesterol (+8.7%) compared with placebo + metformin. Mean percentage increases were noted in low-density lipoprotein cholesterol levels (7.7%, pioglitazone + metformin; 11.9%, placebo + metformin) and total cholesterol (4.1%, pioglitazone + metformin; 1.1%, placebo + metformin), with no significant differences between groups. In the extension study, patients treated with open-label pioglitazone + metformin for 72 weeks had mean changes from baseline of -1.36% in HbA1c and -63.0 mg/dl in fasting plasma glucose. In this study, there was no evidence of drug-induced hepatotoxicity.
The efficacy and tolerability of pioglitazone in combination with a sulfonylurea has been also assessed in patients with type 2 diabetes mellitus. and others . Twenty-three diabetic patients treated with a stable dose of sulfonylurea were randomly assigned to receive either placebo (n = 11) or pioglitazone (45 mg/day) (n = 12) for 16 weeks . Before and after 16 weeks of treatment, all subjects received a 75-g oral glucose tolerance test (OGTT) and peripheral insulin sensitivity was measured with a two-step euglycemic insulin clamp. After 16 weeks pioglitazone treatment significantly decreased fasting plasma glucose, mean plasma glucose during OGTT, and HbA1c without changing fasting or glucose-stimulated insulin/C-peptide concentrations. Fasting plasma free fatty acid (FFA) and mean plasma FFA during OGTT also decreased significantly after pioglitazone treatment. Pioglitazone treatment significantly decreased endogenous glucose production, whereas insulin-stimulated total and non-oxidative glucose disposal was significantly increased indicative of an improvement in hepatic and peripheral (muscle) tissue sensitivity to insulin. Subsequent work has indicated that pioglitazone at doses of 30 and 45 mg/day (but not at doses of 7.5 or 15 mg/day) improves β-cell function along with whole-body insulin sensitivity .
Side Effects
The major side effects of this class of drugs are edema, weight gain, decreased hematocrit and hemoglobin, and elevated (but reversible) alanine aminotransferase activity. Unlike troglitazone, idiosyncratic hepatic reaction does not appear to be a problem with rosiglitazone or pioglitazone. The edema ranges from bothersome trace to anasarca. The mechanism of the edema production is not known. Clinically, diuretics have minimal effect on reducing the edema, though spironolactone may have more benefit than other diuretics. While there are no published studies on this subject, it does appear that the edema is dose dependent. Weight gain may be a modest 2-4 pounds to >20 lbs . Due to their mechanism of action, the risk of hypoglycemia with rosiglitazone or pioglitazone monotherapy is low. Mild to moderate hypoglycemia has been reported during combination therapy with sulfonylureas or insulin .
Cardiovascular Disease
A general listing of the cardiovascular effectsof thiazolidinediones is shown below in Table .
Table Effects of Thiazolidinediones (pioglitazone/rosiglitazone) on Cardiovascular Risk Factors
| Lipid profile |
↑ LDL cholesterol particle size
↑ Lipoprotein a
↑ HDL cholesterol
↓/-- LDL/HDL ratio
↓/↑ Triglycerides
|
| Coagulation and fibrinolysis |
↓ Fibrinogen
↓ Platelet aggregation
|
|
Hemodynamic and vascular
parameters
|
↑ Intravascular volume
↓Blood pressure
↓ Intima-media thickness
↑ Endothelial function
↓ Urine albumin excretion
|
| Inflamatory Markers |
↓ C-reactive protein
↓ Adiponectin
↓ Tumor necrosis factor-α
|
In a meta-analysis by Nissen and Wolski, published literature and clinical trial registries were searched for cardiovascular end points such as MI and death from cardiovascular causes. Data were combined by means of a fixed-effect model. Forty-two trials were selected. In the rosiglitazone group, as compared with the control group, the odds ratio (OR) for MI was 1.43 (95% CI, 1.03-1.98; P = .03), and the OR for death from cardiovascular causes was 1.64 (95% CI, 0.98-2.74; P = .06). The authors concluded that rosiglitazone was associated with a significant increase in the risk of MI and with an increased risk of death from cardiovascular causes that had borderline significance. There were significant limitations to the study including lack of access to original data, which would have enabled time-to-event analysis, no confirmation of events, many studies had only zero or 1 report, and these trials were not designed to capture or adjudicate events. With correction of some of these limitations, another Meta analysis failed to show any statistical increased risk. . Since that time there have been multiple meta analyses looking at variations of the same data with variable results.
A meta analysis of cardiovascular events using patient-level data from randomized trials comparing pioglitazone with a range of alternative regimens determined that death, MI, or stroke occurred in 375 of 8,554 patients (4.4%) receiving pioglitazone and 450 of 7,896 patients (5.7%) receiving control therapy (HR, 0.82; 95% CI, 0.72-0.94; P = .005). The authors concluded that treatment with pioglitazone was associated with a significantly lower risk of death, MI, or stroke compared with any of the alternative regimens. Serious heart failure was increased in patients on pioglitazone, although without an associated increase in mortality (HR, 1.41; 95% CI, 1.14-1.76; P = .002).
There have been two prospective, randomized studies specifically looking at cardiovascular outcome and thiazolidinediones. The study looking at pioglitazone (PROactive study) failed to show a significant benefit of treatment on the primary composite end points (hazard ratio [HR], 0.90; 95% confidence interval [CI], 0.80-1.02; P = .095). However, pioglitazone reduced risk for the main secondary end points, including death from any cause, nonfatal MI, and stroke (HR, 0.84; 95% CI, 0.72-0.98; P = .027). Addition of rosiglitazone to glucose-lowering therapy (RECORD Study) was confirmed to increase the risk of heart failure, but the data remained inconclusive about any possible effect on myocardial infarction. Rosiglitazone did not increase the risk of overall cardiovascular morbidity or mortality compared with standard glucose-lowering drugs.
Fractures
In 2006, an increased risk of fractures was discovered in subjects receiving rosigliatzone participating in the ADOPT Study (A Diabetes Outcome and Progression Trial). Since then there have been multiple reports on the association of thizolidenediones and fractures with a Meta-analysis of 10 randomized controlled trials (13,715 participants) and from 2 observational studies (31679 participants) showed a significant increased risk of fractures in women (OR 2.23, 95%cl 1.65-3.01; p<.001), but not in men. There has been no explanation for the sex difference, but one may speculate that the age of the men in these studies is generally younger than one expects to see significant risk of fractures. Several studies have shown that bone mineral density declines with use of these medications.
α-Glucosidase Inhibitors
Acarbose (Precose®, Glucobay®) and miglitol (Glycet®) are members of the α-glucosidase inhibitor class of oral anti-hyperglycemic compounds that function by blocking the enzymatic degradation of complex carbohydrates in the small intestine . These compounds lower post-prandial glucose and improve glycemic control without increasing the risk for weight gain or hypoglycemia. Each drug is approved for use in the US as monotherapy, which results in a significant reduction in fasting plasma glucose by 25-30 mg/dl, post-prandial glucose by 40-50 mg/dl, and HbA1c by 0.7-1.0% . In addition, acarbose is approved for use in combination with insulin, metformin, or a sulfonylurea, and miglitol is approved for use in combination with a sulfonylurea. The effects of these compounds on glycemic control are additive when used in combination, presumably since their mechanism of action is different. Neither drug is approved in the US for use in combination with a meglitinide or thiazolidinedione. α-glucosidase inhibitors are suitable approaches for patients that have mild to moderate hyperglycemia, or those patients prone to hypoglycemia or at risk for lactic acidosis.
Figure Structure of Acarbose

Mechanism of Action
α-Glucosidase inhibitors are competitive, reversible inhibitors of pancreatic α-amylase and membrane-bound intestinal α-glucosidase hydrolase enzymes. Acarbose, the first α-glucosidase inhibitor discovered, is a nitrogen-containing pseudotetrasaccharide (Figure 1.9), while miglitol is a synthetic analog of 1-deoxynojirimycin (Figure 1.10). The mechanism of action of these inhibitors is similar but not identical. They bind competitively to the oligosaccharide binding site of the α-glucosidase enzymes, thereby preventing enzymatic hydrolysis. Acarbose binding affinity for the α-glucosidase enzymes is: glycoamylase > sucrase > maltase > dextranase . Acarbose has little affinity for isomaltase and no affinity for the β-glucosidase enzymes, such as lactase . Miglitol is a more potent inhibitor of sucrase and maltase that acarbose, has no effect on α-amylase, but does inhibit intestinal isomaltose .
Clinical trials conducted to date have established that the antihyperglycemic effectiveness of acarbose and miglitol is less than 50% than that of either sulfonylureas or metformin. When used as monotherapy, acarbose primarily affects post-prandial glucose levels, which is reduced by 40-50 mg/dl after meal . In most studies, α-glucosidase inhibitors have no significant effects on either fasting insulin or whole body insulin sensitivity in patients with type 2 diabetes. However, there is some evidence that acarbose and voglibose, a structural analog of miglitol in clinical development in Japan, reduces post-prandial hyperinsulinemia in glucose intolerant individuals . Some but not all studies have reported small decreases in fasting or post-prandial triglycerides . Since the mechanism of action of α-glucosidase inhibitors is different from other oral agents, their effects on glycemic control are additive when used in combination. As summarized by Lebovitz , addition of acarbose to sulfonylurea therapy decreases HbA1c by 0.85%; addition of acarbose to metformin therapy decreases HbA1c by 0.73%; and addition of acarbose to insulin therapy decreases HbA1c by 0.54%. As for monotherapy, the predominant improvement is on post-prandial hyperglycemia. Treatment with α-glucosidase inhibitors appears to have a lower rate of secondary failures characteristic of sulfonylurea and metformin therapy.
Side Effects
The major side effects of the α-glucosidase inhibitors are related to gastrointestinal disturbances. These occur in approximately 25-30% of diabetic patients, and include flatulence, diarrhea, bloating, and abdominal discomfort. These side effects can often be minimized by careful dose titration, and sometimes diminish with time. Acarbose is contraindicated in patients with inflammatory bowel disease, cirrhosis, or elevated plasma creatinine (>177 μmol/l). This class of drugs is associated with dose-dependent hepatotoxicity, and serum transaminase levels require monitoring for patients receiving high doses (>200 mg three times daily). Transaminase elevations, which are often asymptomatic, are reversible upon cessation of treatment. Hypoglycemia does not occur in patients on α-glucosidase inhibitor monotherapy. If hypoglycemia occurs while a patient is taking an α-glucosidase inhibitor simultaneously with a sulfonylurea, insulin or a meglitinide, the recommended action is oral administration of pure glucose, dextrose or milk.
INCRETINS
The incretin effect, defined by a significantly greater insulin stimulatory effect evoked after an oral glucose load than that evoked from an intravenous glucose infusion when plasma glucose concentrations are matched, was first described in the 1960’s. This effect is shown below in figure __.
Figure The difference between oral and intravenous glucose on insulin release. There is a significant
increase in insulin with oral vs intravenous glucose - the "incretin effect."

Although other hormones may take part in the incretin effect, the majority of the effect is thought to be due to glucose-dependent insulinotropic peptide (GIP) and glucagon like peptide-1 (GLP-1). The physiological importance of GIP and GLP-1 in overall glucose metabolism has been demonstrated using receptor-knockout animal models,as well as with the use of receptor antagonists. Patients with type 2 diabetes have a significant reduction of the incretin effect, implying that these patients either have decreased concentration of the incretin hormones, or a resistance to their effects. GLP-1 concentrations are reduced in patients with type 2 diabetes in response to a meal, while GIP concentrations are either normal or increased, suggesting a resistance to the actions of GIP thus making GLP-1 a more logical target for therapeutic intervention.
Brown and colleagues isolated the first incretin from cholesystokinin in 1971, and named it gastric inhibitory peptide (GIP). After demonstrating its insulinotrophic properties, Dupre renamed the peptide glucose-dependent insulinotropic peptide (GIP), thus preserving the acronym. Within minutes after ingestion of food, GIP is secreted from the K-cells located in the proximal region of the jejunum. GIP helps maintain normal glucose homeostasis in rodent models, and has an insulinotropic effect in response to hyperglycemia in both animals and humans. However, GIP does not inhibit glucagon secretion, and in fact may stimulate it during euglycemic states, and has no effect on gastric emptying. Furthermore GIP concentrations in patients with type 2 diabetes are either normal, or slightly increased in response to a meal. In patients with type 2 diabetes, GIP infusion has not been able to reduce plasma glucose concentrations, due to a lack of amplification of late phase insulin response to glucose, compared to GLP-1. Thus, GIP has not been considered a suitable candidate for therapeutic development for the treatment of type 2 diabetes.
GLP-1 is cleaved from the proglucagon molecule by the gut specific prohormone convertase enzymes 1 and 3. Two forms of GLP-1 are secreted, GLP-1(7-37) and GLP-1(7-36)amide. The majority of circulating active GLP-1 appears to be GLP-1(7-36)amide. GLP-1 is stored in the L-cells of the ileum and colon, and released at mealtime in response to neurohormonal signals and the presence of food in the gut. GLP-1 exerts its effect on postprandial glucose concentrations through several mechanisms, including enhancing insulin secretion and suppressing postprandial glucagon secretion in a glucose-dependent manner. In addition GLP-1 slows the rate of gastric emptying, which is often paradoxically accelerated in patients with diabetes. GLP-1 also acts as a postprandial satiety signal through neurohormonal networks that signal the brain to suppress appetite and food intake. Furthermore, GLP-1 also has direct effects on the β-cells, as shown in studies done in animal models and cell lines, promoting cell proliferation and neogenesis, while preventing β-cell apoptosis. Additionally, GLP-1 can promote transformation of noninsulin-producing pancreatic cells into cells capable of synthesizing and secreting insulin.
Figure

Figure

GLP-1 reportedly has effects on the liver, skeletal muscle as well as the adipose tissue, resulting in increased glycogen synthesis and peripheral insulin-stimulated glucose uptake. However, this effect is considered controversial, as there have been contradictory observations as well. GLP-1 receptors are also located in the heart, and have been associated with cardiovascular response to stress. Unfortunately in spite of all the potentially beneficial effects of GLP-1 on glucose homeostasis, the therapeutic potential of naturally occurring GLP-1 is limited, due to its extremely short half-life of less than 2 minutes largely due to the degradation by the enzyme dipeptidyl peptidase IV (DPP-IV).
Administration of exogenous GLP-1, as a continuous subcutaneous or intravenous infusion, has been shown to increase insulin secretion, and normalize both fasting and postprandial blood glucose concentrations. As shown in Figure below, an infusion of GLP-1 in a hyperlgycemic Type 2 paient will reduce the glucose level, increase insulin levels and suppress glucagon production.
Figure Glucose-Dependent Effects of GLP-1 on Insulin and Glucagon Levels in Patients With Type 2 Diabetes

In one of the earliest studies with GLP-1, Gutniak et al. showed that a continuous intravenous infusion of GLP-1 resulted in a reduction in the amount of insulin needed to maintain isoglycemia in patients with either type 1 or type 2 diabetes. Few years later, Zander et al., in a non-randomized parallel group study, showed that six weeks of continuous subcutaneous infusion of GLP-1 not only reduced fasting and postprandial plasma glucose concentrations, but also lowered glycosylated hemoglobin (A1C) by 1.3%, and induced up to a 2 kg reduction in body weight.
Juntti-Berggren et al. reported the first randomized trial with the addition of subcutaneous infusion of GLP-1 to regular insulin therapy, and compared it with insulin therapy alone in 12 patients with type 2 diabetes. After initial intensive insulin therapy for 1 week, 8 patients were randomized to receive a subcutaneous infusion of GLP-1 along with injections of regular insulin with meals, and NPH insulin at bedtime, whereas 4 patients were randomized to regular insulin with meals and NPH insulin at bedtime. A significant reduction in postprandial glucose concentration was observed after one week of treatment in patients receiving subcutaneous infusion of GLP-1 added to insulin therapy, compared to insulin therapy alone. Furthermore, an additive lipid-lowering effect was also observed in the GLP-1 infused patients.
In 2001, Larsen et al. reported a randomized trial with continuous intravenous infusion of GLP-1 versus placebo infusion, in 40 hospitalized patients with type 2 diabetes, poorly controlled with sulfonylurea treatment.63 Patients received an infusion of GLP-1 at 4 or 8 ng/kg/min for 16 or 24 hours, versus placebo, for 7 days. Patients infused with GLP-1 showed a significant, dose-dependent, reduction in mean 24-hour plasma glucose concentrations compared with placebo. The fasting and nocturnal plasma glucose concentration was higher in the 16-h GLP-1 infused patients compared to the 24-h GLP-1 infused patients. GLP-1 infusion was also associated with a dose related increase in nausea, headache, and vomiting.
In another randomized study published in 2003 (Meneilly et al.), 16 elderly, insulin-naïve patients with diabetes were divided in two groups of 8 patients each, and given either a continuous subcutaneous infusion of GLP-1 at an initial dose of 100 pmol/kg/h, titrated up to a maximum dose of 120 pmol/kg/h for 12 weeks, compared to controls treated with conventional therapy. Concomitant treatment with glucose lowering agents was discontinued for 1 week prior to study treatment in the group of patients infused with GLP-1, while the control group continued with their usual glucose lowering therapy without any dose changes. GLP-1 infusion was well tolerated, without significant side effects. Hypoglycemia occurred only once in the GLP-1 treatment group, while hypoglycemic events were frequent in the control group (87 events). No significant changes in A1C were observed, despite a discontinuation of oral hypoglycaemic medications in the GLP-1 treated group. There was an enhanced glucose-induced insulin secretion, as well an in insulin-mediated glucose disposal, in the GLP-1 infused group compared to controls.
In 2004, Zander et al. reported a randomized cross-over trial that evaluated the additive effect of continuous infusion of GLP-1 and a TZD, pioglitazone, in patients with type 2 diabetes. Eight patients with type 2 diabetes were given a saline infusion, continuous infusion of GLP-1 (4.8 pmol/kg/min), pioglitazone treatment (30 mg), and a combination treatment with GLP-1 infusion and pioglitazone, in a random order. During the studies with either saline or GLP-1 only infusions, the patients discontinued their normal medications for three weeks, with a 2-day washout period between the saline or GLP-1 infusions. During the studies with pioglitazone with or without GLP-1 infusion, all patients received 30 mg of pioglitazone for 12 weeks and then were randomly assigned to a 48-hour monotherapy period with either pioglitazone or a combination of GLP-1 with pioglitazone. A significant drop in fasting plasma glucose (FPG) concentrations in the GLP-1 only, pioglitazone only, and the combination of pioglitazone with GLP-1 group were observed compared to saline infusion group. FPG was lower in combination therapy compared to monotherapy with either agent. Mean insulin concentrations were significantly higher with GLP-1 compared with pioglitazone. Glucagon concentrations were reduced in both GLP-1 treated arms compared to saline and pioglitazone treatments, and a sensation of appetite reduction was also noted in the GLP-1 treatment arms.
All these studies demonstrated the clinical potential of GLP-1 therapy in patients with type 2 diabetes. Unfortunately, for reasons of practicality, intravenous or subcutaneous continuous infusion of GLP-1 is not practical, as a long-term treatment of diabetes.
Two classes of agents have been developed to take advantage of the unique glucoregulatory potential of GLP-1:
-
The incretin mimetics, such as exenatide (synthetic exendin-4, a peptide resistant to proteolytic cleavage by DPP-IV) or derivatives of GLP-1 (created by chemical modification of the native hormone, to render them resistant to rapid proteolytic cleavage by DPP-IV), targeting the pharmacological effects, and insufficient secretion of GLP-1 seen in type 2 diabetes; and
-
The DPP-IV inhibitors, compounds that increase the concentration of endogenous incretins, including GLP-1, by limiting the proteolytic cleavage by DPP-IV.
Figure Pharmacologic effects of GLP-1

Incretin Mimetics
Exenatide
Exenatide (Byetta) is a synthetic exendin-based GLP-1 analog that 53% homologous to native GLP-1. It is a synthetic version of exendin-4, a peptide originally identified in the saliva of the Gila monster (Heloderma suspectum), and secreted upon ingestion of nutrients. It received regulatory approval in 2005. Exenatide is administered via a pre-filled subcutaneous injection device at a dose of 5 or 10 mcg twice daily, within 60 minutes before morning and evening Current approved indications are add on to metformin, sulfonylurea, thiazolidinedione, a combination of metformin and a sulfonylurea, or a combination of metformin and a thiazolidinedione. It is not currently approved as as monotherapy.
Mechanism of Action
While exendin-4 has many of the properties of GLP-1, the two peptides are unique and are transcribed from distinct genes in the Gila monster. In humans and animal studies, exenatide enhances glucose-dependent insulin secretion, suppresses the elevated postprandial glucagon levels seen in type 2 diabetes, and slows the rate of gastric emptying (which can be paradoxically accelerated in many people with diabetes). In addition, both exenatide and GLP-1 have been reported to promote β-cell proliferation and neogenesis from ductal precursor in animal models. Data obtained in animal models also indicate that exenatide reduces food intake, promotes weight loss, and partially due to this weight loss has an insulin sensitizing effect. Moreover, exenatide has been shown to reduce food intake in healthy humans. Exenatide does not appear to be significantly degraded in the circulation, and is primarily cleared by the kidneys. After a subcutaneous injection, exenatide plasma concentrations increase in a dose-dependent manner (tmax of approximately 2 hours) and decay in a linear manner (t1/2 of 3.3 to 4.0 hours).
Given the mechanism of action, the expected effect of the medication would be to lower postprandial glucose excursions. As shown in a study comparing the addition of either basal insulin or exenatide, exenatide supresses the postprandial excursion while the basal insulin lowers fasting glucoses without any effect on postprandial levels. This is seen in figure below.
Figure Comparison of exenatide vs basal insulin on 24 hour glucose pattern.

Efficacy
Three large randomized, placebo-controlled clinical trials with subcutaneous administration of exenatide (bid), conducted in subjects with type 2 diabetes who were unable to achieve glycemic control with sulfonylureas and/or metformin, demonstrated that exenatide treatment resulted in mean reductions in A1C from baseline of ~1% accompanied by an average weight loss of 2 to 3 kg in those treated with 10 mcg exenatide BID.
Though not approved for monotherapy in a 30-week study of exenatide 10 mcg twice daily, used as monotherapy or in combination with one or two oral therapies there was an A1C reduction of 1.5% from baseline.
Side Effects
Mild-to-moderate nausea was the most commonly reported adverse event associated with exenatide, occurring with greatest frequency upon initiation, and generally subsiding with continued exposure to exenatide.Few new episodes of nausea were reported after 4 weeks of treatment. Furthermore, stepwise dose escalation was found to reduce the incidence of nausea and vomiting. There was no increase in the risk of hypoglycemia when exenatide was administered in combination with metformin, most likely due to the glucose-dependent actions of exenatide. However, the risk of hypoglycemia increased when exenatide was administered with a sulfonylurea, and this risk was greater when A1C was closer to normal, and the dose of sulfonylurea was not concomitantly reduced. The FDA had required addition of two warnings based on post-release reports. In 2008, the product lable was updated to reflect FDA concern over reports of possible association of the drug with pancreatitis In November 2009, based on 78 reports of altered renal function, an alert on possible alteration in function was issued .
Approximately 45% of patients in these studies developed anti-exenatide antibodies; however, the presence of antibodies, and/or the magnitude of the antibody titer, were not associated with an individual patient’s magnitude of glycemic improvement, nor was there an association with incidence of adverse events.
Liraglutide
Liraglutide is a once-daily GLP-1 receptor agonist with 97% amino acid sequence homology to endogenous human GLP-1 approved for clinical use by the FDA in January 2010. It is administered via a pre-filled subcutaneous injection device at a dose of 0.6 mg, 1.2 mg, or 1.8 mg, independently of meals. Current approved indications are as an adjunct to diet and exercise.
Mechanism of Action
Mechanism of action is as described above
Efficacy
Liraglutide at daily subcutaneous monotherapy doses of 1.2 and 1.8 mg as monotherapy decreased HgA1c levels from baseline by –0.84% and –1.14%, respectively, over 52 weeks, achieving significantly greater reductions than sulfonylurea therapy (decrease of of –0.51%). In data from four clinical trials of liraglutide in combination with one or two oral therapies there were reductions of of HgA1c levels of 1.0% to 1.5% for liraglutide 1.2 mg (versus placebo), and –1.0% to –1.5% for 1.8 mg (versus placebo). In these clinical trials of 26 to 52 weeks, liraglutide 1.2 to 1.8 mg/day produced weight reductions of –1.0 to–3.2 kg.
Side Effects
In monotherapy or combination therapy trials, the rate of hypoglycemia ranged from 3% to 12%. There were no episodes requiring assistance from another person. When combinedwith a sulfonylureas the incidence ranged from 5% to 27% with Six liraglutide-treated patients requiring assistance.
5% to 40% of liraglutide-treated patients complained of nausea but this generally subsided within the first 4 weeks of use.
Antibodies to liraglutide were found in 4% to 13% of subjects and did not appear to have any significant effect on the HgA1c-lowering efficacy of liraglutide.
Dipeptidyl Peptidase IV (DPP-IV) Inhibition
As described above, DPP-IV inhibitors are compounds that increase the concentration of endogenous incretins, including GLP-1, by limiting the proteolytic cleavage by DPP-IV. As shown in Figure , above, GLP-1 is an insulinotropic hormone secreted by L-cells of the small intestine that stimulates insulin secretion in a glucose-specific manner, inhibits gastric emptying, suppresses glucagon secretion, and has central anorexic activity . Although it possesses multiple effective clinical activities, administration of GLP-1 is not an ideal approach since it cannot be administered orally. Furthermore, endogenous (and exogenously administered) GLP-1 has undesirable pharmacokinetics; after it is secreted, it is rapidly cleaved and inactivated [plasma half-life < 1 min; ] by the enzyme dipeptidyl peptidase IV (DPP-IV). Thus, inhibition of DPP-IV has been suggested as a feasible alternative to elevate circulating GLP-1 levels, and circumvent the limitations of GLP-1 administration .
The initial, orally active DPP-IV inhibitor, NVP-DPP728, was identified and characterized in vitro and in vivo .
Inhibition of DPP-IV by NVP-DPP728 resulted in a significantly amplified early phase of the insulin response to an oral glucose load in obese fa/fa rats, and restoration of glucose excursions to normal . In contrast, DPP-IV inhibition produced only minor effects in lean FA/? rats. Inactivation of GLP-1 (7-36) amide was completely prevented by DPP-IV inhibition suggesting that the effects of this compound on oral glucose tolerance were mediated by increased circulating concentrations of GLP-1 (7-36) amide.
In DPP-IV(+) [but not in DPP-IV(-)] transgenic rats fed either standard chow or a high-fat diet, NVP-DPP728 significantly suppressed glucose excursions after glucose challenge by inhibiting the plasma DPP-IV activity, associated with the stimulation of early insulin secretion . NVP-DPP728 also improved the glucose tolerance after an oral glucose challenge by potentiating the early insulin response by inhibition of plasma DPP-IV activity in aged DPP-IV(+) Wistar and F344 rats . In contrast, NVP-DPP728 did not affect the glucose tolerance after an oral glucose challenge in aged DPP-IV(-)F344 rats (. Taken together, these results indicate that treatment with NVP-DPP728 ameliorates glucose tolerance in vivo by the direct inhibition of plasma DPP-IV activity, and presumably the subsequent increase in endogenous GLP-1 action.
The clinical activity of this compound as a monotherapy was reported in patients at an early stage of type 2 diabetes . Compared with placebo, NVP-DPP728 at 100 mg (tid; n =31) significantly reduced fasting glucose by 18 mg/dl (1.0 mmol/l), prandial glucose excursions by 21.6 mg/dl (1.2 mmol/l), and mean 24-h glucose levels by 18 mg/dl (1.0 mmol/l). Similar reductions were seen in the 150-mg (bid; n = 32) treatment group. Mean 24-h insulin was significantly reduced by 26 pmol/l in both groups. In the combined active treatment groups, HbA1c was significantly reduced from 7.4 ± 0.7% to 6.9 ± 0.7%. Laboratory safety and tolerability were good in all groups. These results provided clinical proof of concept that inhibition of DPP-IV is a feasible approach for the treatment of type 2 diabetes in the early stage of the disease.
Vildagliptin
Sitagliptin
Sitagliptin ( Januvia™), a selective DPP-IV inhibitor, received regulatory approval in the US in 2006.
Figure Structure of Sitagliptin

Mechanism of Action
As described above, DPP-IV inhibitors are compounds that increase the concentration of endogenous incretins, including GLP-1, by limiting the proteolytic cleavage by DPP-IV. The clinical effect is to stimulate insulin secretion in a glucose-specific manner and suppress glucagon secretion.
Efficacy
In sitagliptin monotherapy studies HgA1c was reduced from baseline by .28% to 0.76% at 12 to 24 weeks. . As an add on agent, there were HgA1c reductions of 0.45% to 1.0% at 24 to 30 weeks. In the studies discussed above, there were no significant effects on weight reported.
Side Effects
As monotherapy, the incidence of hypoglycemic was reported between 0.5% to 2.2%, with no severe episodes. When used in combination with a sulfonylurea,hypoglycemia occurred in 12.2% of patients, but no severe events were reported.
Gastrointestinal events rarely occur. Headaches (incidence of 1.8% to 5.7%), nasopharyngitis (2.9% to 9.1%), and upper respiratory tract infection (0% to8.8%) have been reported.
Saxagliptine
Saxagliptin ( Ongyza), a selective DPP-IV inhibitor, received regulatory approval in the US in 2009.
Figure Structure of Saxagliptine

Mechanism of Action
As described above, DPP-IV inhibitors are compounds that increase the concentration of endogenous incretins, including GLP-1, by limiting the proteolytic cleavage by DPP-IV. The clinical effect is to stimulate insulin secretion in a glucose-specific manner and suppress glucagon secretion.
Efficacy
When added to a sulfonylurea, Saxagliptin at doses of 2.5 mg and 5 mg, decreased HgA1c by 0.54% to 0.64%. The same doses decreased HgA1c by 0.66% to 0.94% when added to a thiazolidinedione (0.30% decrease seen with a thiazolidinedione plus placebo).
Side Effects
At doses of 2.5 and 5 mg, saxagliptin was associated with a hypoglycmia incidence of 2.7% to 4.1% when combined with a thiazolidinedione and 13.3% to 14.6% when combined with a sulfonylurea.).
Bile Acid Sequestrant
Mechanism of Action
Colesevelam hydrochloride (WelCHOL) is a non-absorbed, polymeric, lipid-lowering and glucose-lowering agent intended for oral administration. It is excreted pirmarily in the feces. Colesevelam hydrochloride is a high-capacity bile acid-binding molecule. It is poly(allylamine hydrochloride) cross-linked with epichlorohydrin and alkylated with 1-bromodecane and (6-bromohexyl)-trimethylammonium bromide. The actual glucose lowering mechanism of action remains under investigation. Proposed mechanisms include:
- decreased or slowed absorption of glucose and carbohydrates, reducing postprandial glucose excursions
- alteration of nuclear receptors such as the liver X receptor (LXR).
- alteration of GLP-1 levels
Figure Structure of Colesevelam

Efficacy
Prior to FDA approval, three phase 3 clinical trials investigated the glucose-lowering efficacy of colesevelam HCl when added to insulin-, metformin-, or sulfonylurea based therapy, in patients with inadequately controlled type 2 diabetes (initial HgA1C, 7.5% to 9.5%) . Results showed the addition of colesevelam HCl to the existing, antidiabetes therapy resulted in a significant placebo- corrected reduction in A1C (0.50%, 0.54%, and 0.54%, respectively [P<.001 for all)
Side Effects
As colesevelam is minimaly absorbed from the gastrointestinal tract, few systemic systemic adverse events occur after oral administration. A combination of 4 studies with enrolment of 1129 subjects (567 patients receiving colesevelam; 562 patients receiving placebo) showed 8.6% Colesevelam vs 2.0% placebo subjects complained of constipation. There were no differences in nasopharyngitis, dyspepsia, hypoglycemia, nausea, or hypertension.
Dopamine Agonist
In 2009, bromocriptine mesylate a medication available for many decades, was approved as an adjunct to diet and exercise to improve glycemic control in adults with type-2 diabetes mellitus. This drug specifically approved was a quick-release preparation developed by Vero Science Inc, under the trade name Cycloset. Bromocriptine, is a centrally-acting dopamine D 2 receptor agonist that had been approved for the treatment of hyperprolactinemia-associated dysfunctions, acromegaly and Parkinson disease. The idea of using bromocriptine for the treatment of type-2 diabetes came while studying the metabolism of migrating birds; that they develop seasonal insulin resistance and dopamine plays a role in it.
Mechanism of Action
Based on insulin-glucose clamp studies, the beneficial effect of bromocriptine on glucose homeostasis does not appear to be related to enhanced insulin-mediated glucose disposal or a reduction in basal endogenous glucose production . Potential unexplored mechanisms by which bromocriptine reduces the mean daylong plasma glucose levels include enhanced suppression of endogenous glucose production and/or increased splanchnic glucose uptake after glucose ingestion.
Figure Structure of Bromocriptine

Efficacy
As shown in Figure below, an early short placebo control study demonstrated a modest improvement in fasting glucose and HgA1c levels. In studies filed with the FDA but not yet published, for the specific preparation, Cycloset, demonstrate minimal change in FBS and HgA1c from baseline and modest improvement from placebo. These results are shown in the Table below.
Table Changes in HgA1c and FBS with the Bromocriptine preparation Cycloset
| |
Change from Baseline |
Change from Placebo |
| Monotherapy |
|
|
| HgA1c |
-0.1 |
-0.4 |
| FBS |
0 |
-23 |
| Add to Sulphonylurea |
|
|
| HgA1c |
-0.25 |
-0.55 |
| FBS |
+7 |
-19 |
| Add to metformin and sulphonylurea |
|
|
| HgA1c |
-0.5 |
-0.5 |
Figure Time course of change from baseline in HbA1c and FPG concentrations
in bromocriptine and placebo-treated subjects

Side Effects
The most common adverse events associated with bromocriptine mesylate are nausea, fatigue, dizziness, vomiting and headache. Clinical trial data reveal that bromocriptine mesylate at doses up to 4.8 mg per day was not associated with a different rate of all-cause adverse events compared with placebo. However, the drug is known to be associated with Nasal Stuffiness, Nausea, headache, constrictive pericarditis, neuroleptic malignant syndrome, and hypotension The incidence of hypoglycemia was 6.9% among bromocriptine mesylate-treated patients compared with 5.3% of patients receiving placebo.
In the pooled CYCLOSET phase 3 clinical trials (CYCLOSET N = 2298; placebo N = 1266), adverse events leading to discontinuation occurred in 539 (24%) CYCLOSET-treated patients and 118 (9%) placebo-treated patients. This between-group difference was driven mostly by gastrointestinal adverse events, particularly nausea.
The drug is contraindicated in patients with known hypersensitivity to bromocriptine or ergot-related drugs. It is also contraindicated in patients with syncopal migraine. Bromocriptine increases the likelihood of a hypotensive episode among patients with syncopal migraine. It is also contraindicated in nursing women as it may inhibit lactation. There are postmarketing reports of stroke in these patients although causality has not been proven.
Pramlintide
The hormone amylin (figure 6) is cosecreted with insulin by the pancreatic beta-cells in response to nutrient stimuli. Patients with type 1 diabetes may develop an absolute deficiency of both insulin and amylin, and those with type 2 DM have impaired beta-cell secretion amylin in response to a meal.
Amylin suppresses postprandial arginine-induced glucagon secretion and slowed gastric emptying time. Pramlintide (Figure 6) is a Soluble Analog of Human Amylin allowing clinical application of this hormone.
Figure 6. Structure of Human Amylin and Pramlintide

Mechanism of Action
Pramlintide, an injectable synthetic analog of amylin, slows gastric emptying, attenuates postprandial glucagon secretion, enhances satiety, and reduces food intake.
Efficacy
When added to preprandial insulin, pramlintide improves post-prandial glucose control and promotes weight loss in patients with both type 1 and type 2 diabetes. As shown in figure 7, postprandial glucose excursions are significantly blunted with the addition of pramlitide. Effects on HgA1c reduction are modest, with 52 week net reduction of .27% at 1 year . Similar reductions in hemoglobin A1c and weight have been reported in insulin treated patients with type 2 diabetes.
Figure 7. Effect of Pramlintide on postrandial glucose.

Pramlitide has also been used as a premeal injection without concurrent premeal insulin in Type 2 diabetes. In preliminary studies, when compared to patients receiving preprandial insulin, these patients were able to achieve similar glucose control with less hypoglycemia and without the weight gain seen with preprandial insulin.
Figure 8. Comparison of postprandial insulin and pramlintide on glucoses and weight.

Side Effects
To date, there have not been any significant reports of toxicity to any major organs, significant elevation in laboratory test, alteration in vital signs, electrocardiographic parameters, or physical exams. Overall, nausea, vomiting, and anorexia were the most frequently reported adverse drug events associated with pramlintide therapy. Rates of nausea were generally16.5- 25% (pramlintide) vs 9.5- 16% ( control group). In patients with type 1 DM these symptoms were mild to moderate and transient (within the first 4-8 weeks). The findings were similar in patients with type 1 and those with type 2 DM.
Emerging Approaches
The development pipeline for new oral therapeutic agents for type 2 diabetes is encouraging and continues to expand. These intensive research and development efforts are in response to the increasing prevalence of the disease and related co-morbidities, realization by care givers that successful glycemic control will likely require combination therapy, a growing understanding of the pathophysiology of the disease, and the identification and validation of new pharmacological targets. These targets include receptors and enzymes that: enhance glucose-stimulated insulin secretion, suppress hepatic glucose production, increase skeletal muscle glucose transport and utilization, increase insulin sensitivity and intracellular insulin signaling, and reduce circulating and intracellular lipids (109-113). Due to their promise for future clinical success and because they exhibit mechanisms of action distinct from current therapies, two such emerging approaches will be highlighted here.
Selective Cannabinoid-1 Receptor (CB-1) Antagonism
The appetite-stimulating actitivity of Cannabis sativa has long been known through traditional recreational and medicinal use . The active molecule, delta-9-tetrahydrocannabinol (THC), was initially purified in 1964 , but the discoveries of cannabinoid receptors and their endogenous ligands did not occur until more recently, within the last 15 years . The endocannabinoid system is composed of two distinct G-protein-coupled cannabinoid receptors (CB1 and CB2), two major endogenous ligands (anandamide and 2-arachidonyl glycerol), and the enzymes responsible for ligand synthesis (N-acylphosphatidylethanolamine-specific phospholipase D and diacylglycerol lipase) and degradation (fatty acid amide hydrolase and monoacylglycerol lipase). CB1 receptors are widely distributed on central and peripheral neurons, and also present in adipose tissue, liver, muscle, pancreas, heart, vasculature, and the gastrointestinal tract . The distribution of CB2 receptors is more limited, where they are mainly associated with the immune system and hematopoisesis . Ligand stimulation of the receptors results in decreased adenylate cyclase activity and cAMP production, along with a reduction in intracellular calcium.
The endocannabinoid system was identified as playing a significant role in the regulation of food intake and energy metabolism . There is some evidence that the endocannabinoid system is consituitively active in certain regions of the brain that are known to control energy balance, suggesting that tonic activation favors energy intake, storage, and weight gain . In rodents, activation of CB1 (but not CB2) receptors by endogenous ligands or THC stimulates feeding , while CB1-deficient mice are protected against diet-induced obesity . In human obesity, it has been reported that fatty acid amide hydrolase, the enzyme that degrades anandamide, is reduced , and that a missense mutation is associated with increased body mass index . Taken together, these data have provided the scientific rationale for the idea that pharmacological antagonists of CB1 might offer a promising approach to treat obesity.
Rimonabant is a selective CB1 receptor antagonist for obesity and smoking cessation . Animal studies have convincingly demonstrated that this compound exerts a beneficial effect on reducing hunger, caloric intake, and body weight, and for increasing satiety . In humans, rimonabant was equally effective as judged by initial clinical studies , but due to concern over its depressive side effects, the drug was not approved for use in the United States and after initial approval in Europe was removed from the market. Nevertheless, this mechanism of action may be utilized in the future.
Despite the magnitude of the disease, the choice of oral antihyperglycemic drugs for type 2 diabetes was limited to sulfonylureas for over 40 years. The last 11 years have witnessed the introduction of five new classes of oral antihyperglycemic therapies. Each possesses a distinct mechanism of action, which enables their use independently and, in some cases, as combination therapy. This is important since most patients with type 2 diabetes will require combination therapy to reach an acceptable level of glycemic control . However, despite these advances, there is still plenty of room and necessity for improvement.
The successful long-term management (and hopefully prevention) of type 2 diabetes and its related co-morbidities undoubtedly requires an aggressive, comprehensive approach. This includes intervention at the pre-diabetes stage (e.g. obesity, impaired glucose tolerance) including both changes in lifestyle (i.e. dietary modification and exercise), along with pharmacological intervention that might delay or even prevent the development of the disease and / or its complications. However, once the disease is established and beyond the control of lifestyle modifications, treatment must be initiated and carefully monitored using existing drugs that address the current understanding of the pathophysiology: impaired insulin secretion, increased hepatic glucose production, and peripheral tissue insulin resistance. It is anticipated that emerging agents will also have a beneficial impact on these processes, but with greater efficacy and safety due to a higher degree of selectivity for their molecular targets. In addition, new information regarding the biochemistry, cell biology, and pathophysiology of the disease process is rapidly providing additional exciting opportunities for target identification, validation and subsequent drug development.
The biological effects of emerging agents will not be limited to lowering blood glucose. Already, there are some new agents in clinical development that have been designed to afford a combination of benefits, including the reduction of both lipids and glucose. Other new therapies will likely address the emerging belief that there is a significant inflammatory component of the disease. Perhaps, this might be why tight glycemic control alone has experienced limited success at reducing the risk of macrovascular complications of diabetes. Targeting the risk factors for heart disease clearly requires serious attention and concomitant therapy. The complexity of type 2 diabetes and associated co-morbidities will continue to present a formidable challenge for successful pharmacological treatment. However, based on the growing sophistication of 21st Century research approaches along with the realization of the consequences of failure , there is ample support for the optimistic viewpoint that the current selection of orally active treatment options will continue to expand.
Oral Pharmacological Agents for Type 2 Diabetes
|
|
Tip: To turn text into a link, highlight the text, then click on a page or file from the list above.
|
|
|
|
|


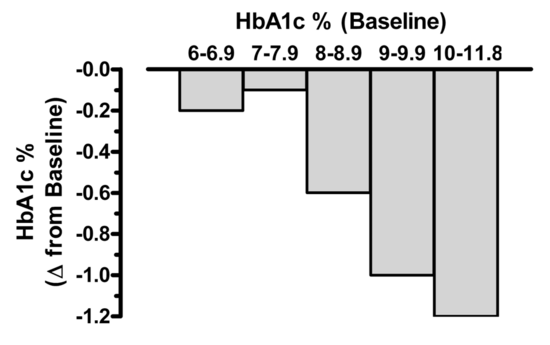


Comments (0)
You don't have permission to comment on this page.