-
If you are citizen of an European Union member nation, you may not use this service unless you are at least 16 years old.
-
You already know Dokkio is an AI-powered assistant to organize & manage your digital files & messages. Very soon, Dokkio will support Outlook as well as One Drive. Check it out today!
| |
Hyperglycemic Crises: Diabetic Ketoacidosis (DKA), And Hyperglycemic Hyperosmolar State (HHS)
Page history
last edited
by Dhemy Padilla 14 years, 6 months ago
HYPERGLYCEMIC CRISES IN PATIENTS WITH DIABETES: DIABETIC KETOACIDOSIS (DKA), AND HYPERGLYCEMIC HYPEROSMOLAR STATE (HHS)
Last Author Revision: 2009
Introduction
Diabetic ketoacidosis (DKA) and hyperosmolar hyperglycemic state (HHS) represent two extremes in the spectrum of marked decompensated diabetes. DKA and HHS are still important causes of morbidity and mortality among patients with diabetes even with major agreements about their diagnostic criteria and treatment protocols . The annual incidence of DKA from population-based studies is estimated to range from 4 to 8 episodes per 1,000 patient admissions with diabetes . The incidence of DKA continues to increase with DKA accounting for about 115,000 hospitalizations in the United States in 2003 (figure 1 a). The rate of hospital admissions for HHS is lower than DKA and is less than 1% of all diabetic-related admissions . Decompensated diabetes imposes a heavy burden in terms of economics and patient outcomes. DKA is an economically burdensome with the average cost of $13,000 per patient per hospitalization .Therefore, the annual expenditure for the care of patients with DKA may exceed $1 billion. The mortality rate for DKA has been falling over the years. Age adjusted mortality rates in the U.S. have dropped by 22% between 1980 and 2001 (from 32 to 20 per 100,000 diabetic population respectively)(4)(figure 1b). Contrary to DKA mortality, the mortality rate of HHS has remained high, ~ 15%, compared to less than 5% in patients with DKA . Severe dehydration, older age, and the presence of comorbid conditions in patients with HHS, account for the higher mortality in these patients .
Figure 1a. Incidence of DKA 1980-2003
Definitions
DKA consists of the biochemical triad of hyperglycemia, ketonemia and metabolic high anion gap acidosis (Figure 2). The terms “hyperglycemic hyperosmolar nonketotic state” and “hyperglycemic hyperosmolar nonketotic coma” have been replaced with the term “hyperglycemic hyperosmolar state” (HHS) to show the facts that 1)the hyperglycemic hyperosmolar state may consist of moderate to variable degrees of clinical ketosis detected by nitroprusside method and 2) alterations in consciousness may often be present without coma .
Both DKA and HHS are characterized by absolute or relative insulinopenia. Clinically, they differ only by the severity of dehydration, ketosis and metabolic acidosis .
DKA most often occurs in patients with type 1 diabetes mellitus (T1DM). It also occurs in type 2 diabetes under conditions of extreme stress such as serious infection, trauma, cardiovascular or other emergencies, and, less often, as a presenting manifestation of type 2 diabetes, a disorder called ketosis-prone type 2 diabetes . Similarly, whereas HHS occurs most commonly in T2DM, it can be seen in T1DM in conjunction with DKA .
Pathogenesis
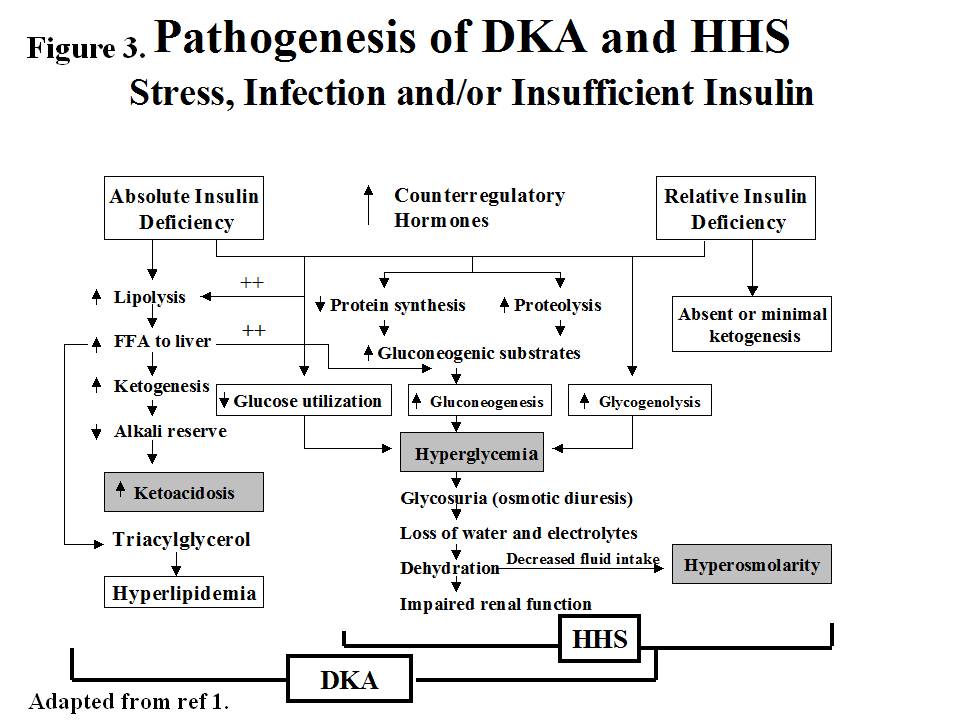
The underlying defects in DKA and HHS are 1) reduced net effective action of circulating insulin as a result of decreased insulin secretion (DKA) or ineffective action of insulin in HHS 2)elevated levels of counterregulatory hormones: glucagon , catecholamines , cortisol , and growth hormone , resulting in increased hepatic glucose production and impaired glucose utilization in peripheral tissues 3)dehydration and electrolytes abnormalities mainly due to osmotic diuresis caused by glycosuria (figure 3).
Figure 3. Pathogenesis of DKA and HHS
In DKA, there is severe alteration of carbohydrate, protein, and lipid metabolism (1). In general, the body is shifted into a major catabolic state with breakdown of glycogen stores, hydrolysis of triglycerides from adipose tissues and mobilization of aminoacids from muscle .The released triglycerides and amino acids from the peripheral tissues will be the substrates for the production of glucose and ketone bodies by the liver .Hyperglycemia and ketone bodies production play central roles in developing this metabolic decompensation .
Hyperglycemia
The hyperglycemia in DKA is the result of three events: (a) increased gluconeogenesis; (b) increased glycogenolysis, and (c) decreased glucose utilization by liver, muscle and fat. Decreased insulin and elevated cortisol levels also result in decreased protein synthesis and increased proteolysis with increased production of amino acids (alanine and glutamine), which serve as substrates for gluconeogenesis . Furthermore, muscle glycogen is catabolized to lactic acid via glycogenolysis. The lactic acid is transported to the liver in the Cori cycle where it serves as carbon skeleton for gluconeogenesis . Increased levels of glucagon, cathecholamines and cortisol with concurrent insulinopenia stimulate gluconeogenic enzymes especially phosphoenol pyruvate carboxykinase (PEPCK) . Decreased glucose utilization is further exaggerated by increased levels of circulating catecholamines and FFA .
Ketogenesis
Excess catecholamines coupled with effective insulinopenia promote triglyceride breakdown (lipolysis) to free fatty acids (FFA) and glycerol, the latter provides carbon skeleton for gluconeogenesis while the former provides the substrate for the formation of ketone bodies . The key regulatory site for fatty acid oxidation is known to be carnitine palmitoyltransferase 1(CPT1)which is inhibited by malonyl CoA in normal nonfasted state but the increase ratio of glucagons and other counter regulatory hormones to insulin disinhibit the fatty acid oxidation and the incoming fatty acids from fat tissue can be converted to ketone bodies . Increased production of ketone bodies (acetoacetate, and ß- hydroxybutyrate) leads to ketonemia . There is also decreased clearance of ketone bodies in DKA, which contributes to ketonemia . These ketoacids are buffered by extracellular and cellular buffers resulting in their loss and subsequent anion gap metabolic acidosis . Studies in diabetic and pancreatectomized patients have demonstrated the cardinal role of hyperglucagonemia and insulinopenia in the genesis of DKA (Figure 4).. In the absence of stressful situations such as dehydration, vomiting or intercurrent illness, ketosis is usually mild .
Elevated levels of pro-inflammatory cytokines and lipid peroxidation markers, as well as procoagulant factors such as plasminogen activator inhibitor-1 (PAI-1) and C-reactive protein (CRP) have been demonstrated in DKA. The levels of these factors return to normal with insulin therapy and correction of hyperglycemia . This inflammatory and procoagulant state may explain the well-known association between hyperglycemic crisis and thrombotic state.
Hyperglycemic Hyperosmolar State
The pathogenesis of DKA and HHS are similar, however, in HHS: 1) there is enough insulin to prevent lipolysis and ketogenesis but not adequate to cause glucose utilization (as it takes 1/10 as much insulin to suppress lipolysis as it does to stimulate glucose utilization)(47,48) 2) possible smaller increases in counterregulatory hormones
Precipitating factors
The two most common precipitating factors in the development of DKA or HHS are inadequate or inappropriate insulin therapy or infection . Other factors include myocardial infarction, cerebrovascular accidents,pulmonary embolism, pancreatitis ,alcohol abuse and drugs (Table 1) . In addition to the mentioned precipitating factors, numerous underlying medical illness and medications that provoke the release of counter regulatory hormones and/or compromise the access to water are likely to result in severe dehydration and HHS . Drugs such as corticosteroids, thiazides, sympathomimetic agents (e.g.,dobutamine and terbutaline ) and second generation antipsychotic agents may precipitate the genesis of DKA or HHS. In young patients with type 1 diabetes, insulin omission due to fear of hypoglycaemia or weight gain, the stress of chronic disease, and eating disorders, may contribute in 20 % of recurrent DKA . Cocaine use also is associated with recurrent DKA . Mechanical problems with continuous subcutaneous insulin infusion (CSII) devices had also precipitated DKA , but with improvement in technology and better education of patients ,the incidence of DKA seems to have been reduced in pump users . Further studies are required to document reduction of DKA incidence with the use of CSII devices. There are also case reports of patients with DKA as the primary manifestation of acromegaly .
Increasing numbers of DKA cases have been reported in patients with DMT2. Available evidence shows that almost 50 % of newly diagnosed adult African American and Hispanic patients with DKA have type 2 diabetes .These patients who have ketosis– prone type 2 diabetes develop sudden-onset impairment in insulin secretion and action, resulting in profound insulinopenia . Clinical and metabolic features of these patients include a high rate of obesity, a strong family history of diabetes, a measurable pancreatic insulin reserve and low prevalence of autoimmune markers of ß-cell destruction .Aggressive management with insulin improves ß cell function , leading to discontinuance of insulin therapy within a few months of follow-up and 40 % of these patients remain non insulin dependent for 10 years after the initial episode of DKA .The etiology of acute transient failure of ß-cells leading to DKA in these patients is not known but the suggested mechanisms include glucotoxicity, lipotoxicity, and genetic predisposition .
A genetic disease, glucose-6-phosphate dehydrogenase deficiency, has been linked with ketosis-prone diabetes .
Table 2 sumarizes the common precipitating factors in DKA.
Table 2. Precipitating factors for DKA

Clinical Features
Symptoms and signs
DKA evolves rapidly within a few hours of the precipitating event(s). On the other hand, development of HHS is insidious and may occur over days or weeks .
The common clinical pictures in DKA and HHS due to hyperglycemia include polyuria, polyphagia, polydipsia, weight loss, weakness and physical signs of dehydration such as dry buccal mucosa, sunken eye balls, poor skin turgor, tachycardia, hypotension and shock in severe cases. Kussmaul respiration, acetone breath, nausea, vomiting and abdominal pain may also occur primarily in DKA . Abdominal pain, which correlates with the severity of acidosis , may be severe enough to be confused with acute abdomen in 50-75% of cases . Therefore, in the presence of acidosis, DKA as an etiology of abdominal pain should be considered . Patients often are normothermic or mild hypothermia, even when harbouring an infection .Therefore careful search for a source of infection should be performed even in the absence of fever . Mental status in DKA may vary from full alertness to profound lethargy or coma but less frequent than HHS . The relationship of depressed consciousness to higher serum osmolality in DKA and its causes has been controversial , some studies have suggested that pH is its cause while others have concluded that osmolality is responsible for the comatose state. More recently it has been proposed that consciousness level in children with DKA is related to the severity of acidosis (pH) but not to blood glucose . This however was reported in children under the age of 16. In our earlier studies of patients with DKA using low dose versus high dose insulin therapy, we evaluated the initial biochemical values of 48 patients with stupor/coma versus non comatose patients . Table 3 shows that only glucose, bicarbonate, BUN and osmolality, but not pH , were significantly different between non comatose and comatose patients. Furthermore in 3 separate studies in which 123 cases of DKA were evaluated, the most significant finding was related to osmolality as demonstrated in figure 5 . This finding was also repeated in other study with different patients which showed osmolality was related significantly to glucose levels but not to pH . Furthermore ,according to a recent study, ICU-admitted patients with DKA are less ill, and have lower disease severity scores, mortality, and shorter length of ICU and hospital stay, than non-DKA patients. Disease severity scores are not, but precipitating cause is, predictive of prolonged hospital stays in patients with DKA
In patients with HHS, final symptoms include clouding of sensorium which progresses to mental obtundation or coma . Occasionally, patients with HHS may present with focal neurological deficit and seizure disorders . Most of the patients with HHS and an effective osmolarity of > 320 mOsm/kg are severely obtundated or comatose, but altered mental status rarely exists in patients with osmolarity of < 320 mOsm/kg (Figure 5).Therefore, severe alteration in the level of consciousness in patients with osmolarity of < 320 mOsm/kg requires evaluation for other causes including CVA and other catastrophic events like myocardial and bowel infarctions .
Laboratory Abnormalities
The initial laboratory evaluation of patients with suspected DKA or HHS should include determination of plasma glucose, blood urea nitrogen, creatinine , serum ketones, electrolytes (with calculated anion gap), osmolality, urinalysis, urine ketones by dipstick, arterial blood gases and complete blood count with differential. An electrocardiogram, blood, urine or sputum cultures and chest X-ray should also be performed, if indicated. HbA1c may be useful in differentiating chronic hyperglycemia of uncontrolled diabetes from acute metabolic decompensation in a previously well controlled diabetic patient . Table 1 summarizes the biochemical criteria for DKA and HHS and electrolyte deficits in these two conditions. It also provides a simple method for calculating anion gap and serum osmolality.
DKA is classified as mild, moderate, or severe based on the severity of metabolic acidosis and the presence of altered mental status . Over 30% of patients have features of both DKA and HHS . Patients with HHS typically have pH > 7.30, bicarbonate level > 20 mEq/L, and negative ketone bodies in plasma and urine. However, some of them may have ketonemia. Some studies on serum osmolarity and mental alteration have established a positive linear relationship between osmolarity and mental obtundation . Therefore , the occurrence of coma in the absence of definitive elevation of serum osmolality requires immediate consideration of other causes of mental status change .
The admission biochemical data in patients with DKA or HHS are shown in Table 4.
Table 4. Admission biochemical data in patients with HHS and DKA.
The major cause of water deficit in DKA and HHS is glucose-mediated osmotic diuresis which leads to loss of water in excess of electrolytes . Despite the excessive water loss, the admission serum sodium tends to be low because serum glucose in the presence of insulinopenia of DKA and HHS cannot penetrate to cells. Therefor it becomes osmotically effective and pulls water from intracellular space to the extra cellular space and dilutes the sodium concentration . True sodium concentration (millimolar )can be obtained by multiplying excess glucose above 100 mg/dl by 1.6 /100 . If the corrected sodium level is extremely low , hypertriglyceridemia (secondary to uncontrolled diabetes ) should be suspected. In this condition the plasma becomes milky and lipema retinalis may be visible in physical examination . Serum potassium may be elevated on arrival due to insulin deficiency, dehydration and a shift of potassium from intracellular to extra cellular compartments in response to acidosis . On occasion, the initial potassium level may be normal or low, which is a danger sign Initiation of therapy, which leads to the transfer of potassium into cells, may cause fatal hypokalemia if potassium is not added early . Phosphate depletion in DKA is universal but on admission, like the potassium, it may be low ,normal or high .
Leukocytosis is a common finding in patients with DKA or HHS , but leukocytosis greater than 25,000 /μL suggests ongoing infection requiring further work up . The exact etiology of this non specific leukocytosis is not known. A recent study also showed non specific leukocytosis in subjects with hyoglycaemia induced by insulin injection and suggested that this phenomenon may be due to the increased levels of cathecholamine, cortisol, and proinflammatory cytokines such as TNF-α during acute stress . Hypertriglyceridemia may be present in HHS and is almost always seen in DKA . Hyperamylasemia, which correlates with pH and serum osmolality and elevated level of lipase may occur in 16 - 25% of patients with DKA . The origin of amylase in DKA is usually non-pancreatic tissue such as the parotid gland .
Pitfalls of Laboratory Tests
False positive values for lipase may also be seen if plasma glycerol levels are very high due to rapid breakdown of adipose tissue triglycerides (glycerol is the product measured in most assays for plasma lipase). Therefore, pancreatic enzymes may not be reliable tools for the diagnosis of pancreatitis in the setting of DKA . Other pitfalls include artificial elevation of serum creatinine, either as a result of dehydration or interference from ketone bodies if a colorimetric method is used . Most of the laboratory tests for ketone bodies use the nitroprusside method, which detects acetoacetate, but not β hydroxybutyrate (BOHB ) . Since BOHB is converted to acetoacetate during treatment , the ketone test may show high values suggesting erroneously that ketonemia is deteriorating, therefore the follow up measurement of ketones during the treatment by nitroprusside method is not recommended .Newer glucose meters have the capability to measure BOHB, which overcomes this problem ,( Figure 6 ). Furthermore drugs that have sulfhydryl groups can interact with the reagent in the nitroprusside reaction, giving a false positive result . Particularly important in this regard is captopril, an angiotensin converting enzyme inhibitor prescribed for the treatment of hypertension and diabetic nephropathy. Therefore for the diagnosis of DKA, clinical judgement and consideration of other biochemical data are required to interpret the value of positive nitroprusside reactions in patients on captopril .
DIFFERENTIAL DIAGNOSIS
Patients may present with metabolic conditions resembling DKA or HHS. For example, in alcoholic ketoacidosis (AKA), total ketone bodies are much greater than in DKA with a higher BOHB to acetoacetate ratio of 7:1 versus a ratio of 3:1 in DKA . The AKA patients seldom present with hyperglycemia . It is also possible that patients with a low intake of food will present with mild ketoacidosis (starvation ketosis) but these patients rarely present with serum bicarbonate concentration less than 18, and do not exhibit hyperglycemia . Additionally, DKA has to be distinguished from other causes of high anion gap acidosis including lactic acidosis, advanced chronic renal failure as well as ingestion of drugs such as salicylate, methanol and ethylene glycol. Isopropyl alcohol, which is commonly available as rubbing alcohol can cause considerable ketosis and high osmolar gap without metabolic acidosis, but it has a tendency to cause hypoglycemia rather than hyperglycemia . Finally, patients with diabetes insipidus presenting with severe polyuria and dehydration, who are treated with dextrose water, can have hyperglycemia ; a clinical picture that can be confused with HHS , (Table 5).
Table 5. Laboratory evaluation of metabolic causes of acidosis and coma
TREATMENT
The goals of therapy in patients with DKA and HHS include 1) Improvement of circulatory volume and tissue perfusion, 2) Gradual reduction of serum glucose and plasma osmolarity, 3) correction of electrolyte imbalance, and in DKA steady resolution of ketosis, and 4) Identification and prompt treatment of co-morbid precipitating causes . It must be emphasized that successful treatment of DKA and HHS requires frequent monitoring of patients regarding the above goals by clinical and laboratory parameters. Protocols for the management of patients with DKA and HHS are illustrated in figures 7 . A flow sheet such as the one shown in figure 8 should be provided for monitoring the patients .
Fluid Therapy
DKA and HHS are volume-depleted states with water deficit of approximately 6 L in DKA and 9 L in HHS . Therefore, the initial fluid therapy is directed toward expansion of interstitial and intravascular volume and securing adequate urine flow . The initial fluid of choice is isotonic saline, which we recommend to be infused at the rate of 15–20 ml /kg body weight per hour or 1–1.5 L during the first hour. The choice of fluid for continued repletion depends on the hydration status, serum electrolyte levels, and urinary output. In patients who are hypernatremic or eunatremic, 0.45% NaCl infused at 4–14 ml/kg/hour is appropriate and in patients with hyponatremia 0.9% NaCl at a similar rate is preferred. The goal is to replace half of the estimated water deficit over a period of 12- 24 hours . In patients with hypotension, aggressive fluid therapy with isotonic saline should continue until blood pressure is stabilized. The administration of insulin without fluid replacement in such patients may further aggravate hypotension . Furthermore, the use of hydrating fluid in the first hour of therapy before insulin has multiple advantages including a) providing a chance to obtain serum potassium value before insulin administration, b) preventing possible deterioration of hypotensive patients with the use of insulin without adequate hydration, and c) increasing insulin effectiveness by decreasing the serum osmolality . Hydration alone may also reduce the level of counterregulatory hormones and hyperglycemia . Hydration reduces serum blood glucose, BUN, and potassium levels without significant changes in pH or HCO3 .The mechanism for lowering glucose is believed to be due to osmotic diuresis and modulation of conterregulatory hormone release . Patients with DKA and HHS require calories for proper metabolism of ketone bodies. Therefore in DKA, as soon as blood glucose falls below 200 mg/dl, the sodium chloride solution should be replaced with 5% glucose containing saline solution with a reduced rate of insulin administration until acidosis and ketosis are controlled while avoiding too rapid correction of hyperglycemia (which may be associated with cerebral edema especially in children) and also inhibiting hypoglycemia . In HHS, the use of D5 ½ NS should start when blood glucose reaches 300 mg/dl, because overzealous replacement with hypotonic fluids has been associated with the development of cerebral edema (109). It should be emphasized that the replacement of urinary losses is also important as failure may lead to delay in the restoration of sodium, potassium, and water deficits .
Insulin Therapy
The cornerstone of DKA and HHS therapy is insulin in physiologic doses . Insulin should not be given to patients unless the serum potassium value is > 3.3 mEq/l . We recommend the use of IV bolus of regular insulin (0.1 U/kg body weight) and continuous infusion of regular insulin at the dose of 0.1U/kg/hr as the method of choice. The optimal rate of glucose reduction would be between 50- 70 mg/hr. If it is not achieved in the first hour, the insulin dose may be doubled in order to obtain constant decline in glucose level. As mentioned earlier, when plasma glucose reaches 200 mg/dl for DKA or 300 for HHS, the hydration fluid should be changed to D5 ½ NS, and insulin rate should be decreased to 0.05 U/kg/hr. The rate of insulin should be adjusted to maintain blood glucose between 150-200 mg/dl in DKA and 250-300 mg/dl for HHS until DKA is resolved or mental obtundation and hyperosmolar state are corrected in HHS .
A study which investigated the optimum route of insulin therapy in DKA demonstrated that the time for resolution of DKA was identical in patients who received regular insulin via intravenous, intramuscular, or subcutaneous routes. However, patients who received intravenous insulin showed a more rapid decline in blood glucose and ketone bodies in the first 2 hours of treatment . Patients who received intravenous insulin, attained an immediate pharmacologic level of insulin concentration. Thus, it was established that an intravenous loading dose of insulin would be beneficial regardless of the subsequent route of administration during treatment. A follow up study demonstrated that a priming or loading dose given half by intravenous route and half by intramuscular route was as effective as one dose given intravenously in lowering the level of ketone bodies in the first hour .
A bolus or priming dose of insulin has been used in a number of studies. The need of such a method, when using intravenous infusion of insulin, is not clear as there is no prospective randomized study to establish efficacy of bolus or priming dose before infusion of insulin. However our study in children demonstrated the effectiveness of intravenous injection of insulin without a bolus dose . In the most recent consensus statement by the American Diabetes Association for treatment of DKA in children, the use of bolus insulin is not recommended . Furthermore, our preliminary study on the use of bolus versus no bolus dose demonstrated no difference between the two methods . Therefore it would appear that if intravenous insulin is used, priming or bolus dose insulin may not be necessary.
Recent clinical studies have shown the potency and cost effectiveness of subcutaneous rapid-acting insulin analogs (lispro or aspart) in the management of patients with uncomplicated mild to moderate DKA . The patients received subcutaneous rapid-acting insulin doses of 0.2 U/kg initially, followed by 0.1 U/kg every 1 hour or an initial dose of 0.3 U/kg followed by 0.2 U/kg every 2 hours until blood glucose was < 250 mg/dl. Then the insulin dose was decreased by half to 0.05, or 0.1 U/kg respectively, and administered every 1 or 2 hours until resolution of DKA. As shown in table 6, there were no differences in length of hospital stay, total amount of insulin needed for resolution of hyperglycemia or ketoacidosis, or in the incidence of hypoglycemia among treatment groups . The use of insulin analogs allowed treatment of DKA in general wards or the emergency department and so reduced cost of hospitalization by 30% without any significant changes in hypoglycaemic events . Similar results has been reported recently in pediatric patients with DKA . It is important to point out that the use of fast-acting insulin analogs is not recommended for patients with severe DKA or HHS, as there are no studies to support their use. Again these agents may not be effective in patients with severe fluid depletion since they are given subcutaneously .
Table 6. Comparative Effects of Subcutaneous (SC) Fast-acting Insulin vs IV Regular Insulin in DKA. (Data Adapted from References ).
|
|
Aspartate *
SC -2hr.
|
Lispro *
SC-1 hr.
|
Regular **
IV.
|
P values
|
|
Length of hospital stay (days)
|
3.9± 1.5
|
4 ± 2
|
4.5 ± 3.0
|
NS
|
|
Duration of therapy until BG<250 mg/dl (hrs)
|
6.1 ± 1
|
7 ± 1
|
7.1 ± 1
|
NS
|
|
Amount of insulin until resolution of DKA
|
10.7 ± 0.8
|
10 ± 1
|
11 ± 0.7
|
NS
|
|
Episodes of hypoglycemia
|
94 ± 32
|
84 ± 32
|
82 ± 28
|
NS
|
|
Episodes of Hypoglycemia
|
1
|
1
|
1
|
NS
|
|
Cost of Hospitalization
|
10,173 ± 1738
|
9,816 ± 4981
|
17,030 ± 1753
|
<0.01
|
Potassium Therapy
Although total-body potassium is depleted , mild to moderate hyperkalemia is frequently seen in patients with DKA, due to acidosis, proteolysis and insulinopenia. Insulin therapy, correction of acidosis, and volume expansion decrease serum potassium concentrations. To prevent hypokalemia, potassium replacement is initiated after serum levels fall below 5.3 mEq/l, in patients with adequate urine output (50 ml/h). Adding 20–30 mEq potassium to each liter of infused fluid is sufficient to maintain a serum potassium concentration within the normal range of 4–5 mEq/L . Patients with DKA who had severe vomiting or had been on diuretics may present with significant hypokalemia. In such cases, potassium replacement should begin with fluid therapy, and insulin treatment should be postponed until potassium concentration becomes > 3.3 mEq/L; in order to prevent arrythmias and respiratory muscle weakness .
Bicarbonate Therapy
The use of bicarbonate in treatment of DKA remains controversial . In patients with pH > 7.0, insulin therapy inhibits lipolysis and also corrects ketoacidosis without use of bicarbonate . Bicarbonate therapy has been associated with some adverse effects, such as hypokalemia , decreased tissue oxygen uptake and cerebral edema and delay in the resolution of ketosis . However, patients with severe DKA (low bicarbonate <10, or Pco2 < 12) may experience deterioration of pH if not treated with bicarbonate . A prospective randomized study in patients with pH between 6.9 and 7.1 showed that bicarbonate therapy had no risk or benefit in DKA . Therefore, in patients with pH between 6.9 and 7.0, it may be beneficial to give 50 mmol of bicarbonate in 200 ml of sterile water with 10 mEq KCL over two hours to maintain the pH at > 7.0. . Considering the adverse effects of severe acidosis such as impaired myocardial contractility, adult patients with pH < 6.9 should be given 100 mmol sodium bicarbonate in 400 ml sterile water (an isotonic solution) with 20 mEq KCl administered at a rate of 200 ml/h for two hours until the venous pH becomes greater than 7.0. Venous pH should be assessed every 2 hours until the pH rises to 7.0; treatment can be repeated every 2 hours if necessary .
Phosphate Therapy
There is no evidence that phosphate therapy is necessary in treatment for better outcome of DKA . However, in patients with potential complications of hypophosphatemia, including cardiac and skeletal muscle weakness, the use of phosphate may be considered . Phosphate administration may result in hypocalcemia when used in high dose .
Treatment of HHS
A similar therapeutic protocol is also recommended for treatment of HHS ,but no bicarbonate therapy is needed for HHS, and changing to glucose-containing fluid is done when blood glucose reaches 300 mg/dl.
Follow-up
During follow up, blood should be drawn every 2–4 h for determination of serum electrolytes, glucose, blood urea nitrogen, creatinine, osmolality, and venous pH. After the initial arterial pH is drawn, venous pH can be used to assess the acid/base status (12). An equivalent arterial pH value is calculated by adding 0.03 to the venous pH value . The resolution of DKA is reached when the blood glucose is < 200 mg/dl, serum bicarbonate is > 18, pH is > 7.30 and anion gap is < 12 . HHS is resolved when osmolarity is < 320 mOsm/kg with a gradual recovery to mental alertness. The latter may take twice as long as to achieve blood glucose control. Ketonemia typically takes longer to clear than hyperglycemia .
Once DKA has resolved, patients who are able to eat can be started on a multiple dose insulin regimen with a long acting insulin and short/rapid acting insulin given before meals as needed to control plasma glucose. Intravenous insulin infusion should be continued for 1–2 h after giving the subcutaneous insulin to maintain adequate plasma insulin levels. Immediate discontinuation of intravenous insulin may lead to hyperglycemia or recurrence of ketoacidosis. If the patient is unable to eat, it is preferable to continue the intravenous insulin infusion and fluid replacement. Patients with known diabetes may be given insulin at the dose they were receiving before the onset of hyperglycaemic crises. In patients with new onset diabetes, a multi-dose insulin regimen should be started at a dose of 0.5-0.8 U/kg per day, including regular or rapid-acting and basal insulin; until an optimal dose is established .
COMPLICATIONS
The most common complications of DKA and HHS include hypoglycemia and hypokalemia due to overzealous treatment with insulin and bicarbonate (hypokalemia), but these complications occur infrequently with current low dose insulin regimens.. During the recovery phase of DKA, patients commonly develop a short-lived hyperchloremic non-anion gap acidosis, which usually has few clinical consequences . Hyperchloremic acidosis is caused by the loss of large amounts of ketoanions; which are usually metabolized to bicarbonate during the evolution of DKA ; and excess infusion of chloride containing fluids during treatment .
Cerebral edema, a frequently fatal complication of DKA, occurs in 0.7–1.0% of children, particularly those with newly diagnosed diabetes . It may also occur in patients with known diabetes and in very young adults usually under 20 years of age . Cerebral edema has also been reported in patients with HHS, with some cases of mortality . Clinically, cerebral edema is characterized by deterioration in the level of consciousness, lethargy, decreased arousal, and headache .Headache is the earliest clinical manifestation of cerebral edema . This is followed by altered level of consciousness and lethargy. Neurological deterioration may lead to seizures, incontinence, pupillary changes, bradycardia, and respiratory arrest. It may be so rapid in onset due to brain stem herniation that no papilledema is found. If deteriorating clinical symptoms occur, the mortality rate may become higher than 70%, with only 7–14% of patients recovering without permanent neurological deficit. Manitol infusion and mechanical ventilation are used to combat cerebral edema. The cause of cerebral edema is not known with certainty. It may result from osmotically driven movement of water into the central nervous system when plasma osmolality declines too rapidly during treatment of DKA or HHS . Although the osmotically mediated mechanism seems plausible, a recent study using magnetic resonance imaging (MRI) showed that cerebral edema was due to increased cerebral perfusion . Another postulated mechanism for cerebral edema in patients with DKA involves the cell membrane Na+/H+ exchangers, which are activated in DKA. The high H+ level allows more influx of Na+ thus increasing more influx of water to the cell with consequent edema . ß-hydoxybutyrate and acetoacetate also play a role in the pathogenesis of cerebral edema . These ketone bodies have been shown to affect vascular integrity and permeability, leading to edema formation . In summary, reasonable precautionary measures to decrease the risk of cerebral edema in high-risk patients include 1) avoidance of overenthusiastic hydration and rapid reduction of plasma osmolality and 2) closed hemodynamic monitoring .
Hypoxemia and rarely non-cardiogenic pulmonary edema may complicate the treatment of DKA . Hypoxemia may be related to the reduction in colloid osmotic pressure that leads to accumulation of water in lungs and decreased lung compliance .The pathogenesis of pulmonary edema may be similar to that of cerebral edema suggesting that the sequestration of fluid in the tissues may be more widespread than is thought. Thrombotic conditions and disseminated intravascular coagulation may contribute to the morbidity and mortality of hyperglycemic emergencies . Prophylactic use of heparin, if there is no gastrointestinal hemorrhage, should be helpful .
PREVENTION
Several studies suggest that the omission of insulin is one of the most common precipitating factors of DKA ,sometimes because patients are socio-economically underprivileged, and may not have access to or afford medical care. In addition, they may have a propensity to use illicit drugs such as cocaine, which has been associated with recurrent DKA . Therefore, it is important that medical care be provided for these patients. Education of the patient about sick day management is very vital to prevent DKA, and should include information on when to contact the health care provider, blood glucose goals, use of insulin and initiation of appropriate nutrition during illness and should be reviewed with patients periodically. Patients must be advised to continue insulin and to seek professional advice early in the course of the illness. Close follow up is very important, as it has been shown that three-monthly visits to the endocrine clinic will reduce the number of ER admission for DKA . Close observation, early detection of symptoms and appropriate medical care would be helpful in preventing HHS in the elderly.
A recent study (150) in adolescents with type 1 diabetes suggests that some of the risk factors for DKA include higher HbA1c, uninsured children and psychological problems . In other recent studies , education of primary care providers and school personnel in identifying the signs and symptoms of DKA has been shown to be effective in decreasing the incidence of DKA at the onset of diabetes . In another study outcome data of 556 patients with diabetes under continuing care over a 7-year period were examined .The hospitalization rates for DKA and amputation were decreased by 69 % due to continuing care and education .
Considering DKA and HHS as potentially fatal and economically burdensome complications of diabetes, every effort for diminishing the possible risk factors is worthwhile.
Hyperglycemic Crises: Diabetic Ketoacidosis (DKA), And Hyperglycemic Hyperosmolar State (HHS)
|
|
Tip: To turn text into a link, highlight the text, then click on a page or file from the list above.
|
|
|
|
|













Comments (0)
You don't have permission to comment on this page.